Therapeutic potential of NAD-boosting molecules: the in vivo evidence
Summary
Nicotinamide adenine dinucleotide (NAD), the cell’s hydrogen carrier for redox enzymes, is well known for its role in redox reactions. More recently, it has emerged as a signaling molecule. By modulating NAD+ sensing enzymes, it controls hundreds of key processes from energy metabolism to cell survival, rising and falling depending on food intake, exercise and the time of day. NAD+ levels steadily decline with age, resulting in altered metabolism and increased disease susceptibility. Restoration of NAD+ levels in old or diseased animals can promote health and extend lifespan, prompting a search for safe and efficacious NAD-boosting molecules. Such molecules hold the promise of increasing the body’s resilience, not just to one disease, but to many, thereby extending healthy human lifespan.
eTOC Blurb
Nicotinamide adenine nucleotide (NAD+) has emerged as a key regulator of cellular processes that control the body’s response to stress. Rajman et al. discuss NAD boosters, small molecules that raise NAD+ levels, which are now considered to be highly promising for the treatment of multiple diseases and the potential extension of human lifespan.
The rise, fall, and rise of NAD
Nicotinamide adenine dinucleotide (NAD) is one of the most important and interesting molecules in the body. It is required for over 500 enzymatic reactions and plays key roles in the regulation of almost all major biological processes (Ansari and Raghava, 2010). Above all, it may allow us to lead healthier and longer lives.
NAD was first described in 1906 by Harden and Young as a cell component that enhanced alcohol fermentation (Harden and Young, 1906). Then in 1936, Warburg showed that NAD is required for redox reactions (Warburg and Christian, 1936) and solidified the nomenclature: “NAD” refers to the chemical backbone irrespective of charge, “NAD+” and “NADH” refer to the oxidized and reduced forms respectively.
By 1960, it was assumed that all the exciting work on NAD had been done. In 1963, a breakthrough came with the discovery that NAD+ is a co-substrate for the addition of poly-ADP-ribose to proteins (Chambon et al., 1963). PARylation, as it is now called, is carried out by the poly(ADP-ribose) polymerases (PARPs), a family of 17 proteins involved in a wide variety of cellular functions. Some PARPs are mono-(ADP-ribose) transferases, so a better name for these proteins is MARTs (Bai, 2015; Gupte et al., 2017). PARPs control numerous cellular functions, from DNA repair to gene expression, though multiple members remain poorly characterized.
Much of the renewed interest in NAD over the last decade can be attributed to the sirtuins, a family of NAD+-dependent protein deacylases (SIRT1-7). In 1999 Frye discovered that mammalian sirtuins metabolized NAD+ (Frye, 1999), then the Guarente and Sternglanz labs revealed that yeast Sir2 is an NAD+-dependent histone deacetylase (Imai et al., 2000; Landry et al., 2000). Since then, sirtuins have been shown to play a major regulatory role in almost all cellular functions. At the physiological level, sirtuins impact inflammation, cell growth, circadian rhythms, energy metabolism, neuronal function, and stress resistance (Gertler and Cohen, 2013; Imai and Yoshino, 2013). In this article, we review the physiology, pharmacology and potential use of “NAD-boosting molecules” for the treatment of diverse diseases and potentially even aging.
NAD+ synthesis
NAD+ is one of the most abundant molecules in the human body, required for approximately 500 different enzymatic reactions and present at about three grams in the average person. Though it was once considered a relatively stable molecule, NAD+ is now known to be in a constant state of synthesis, degradation and recycling, not only in the cytoplasm but also within major organelles including the nucleus, Golgi and peroxisomes (Anderson et al., 2003). Recent advancements in high-resolution, high-sensitivity NAD+ metabolite tracing methods such as mitoPARP, PARAPLAY, and Apollo-NADP+ (Cambronne et al., 2016; Cameron et al., 2016; Dolle et al., 2010) have revealed that the concentration and distribution of NAD+ and its metabolites are different depending on the cell compartment and change in response to physiological stimuli and cellular stress. NAD+ has two main pools, the “free” pool and protein-associated, “bound” pool, and the ratio of these pools varies across different organelles, cell types, tissues and even the age of individuals. There is also evidence that there are rapid, local fluctuations of NAD+ (Zhang et al., 2012).
With the exception of neurons, mammalian cells cannot import NAD+, so they must synthesize it either de novo by the kynurenine pathway from tryptophan (trp), or forms of vitamin-B3 such as nicotinamide (NAM) or nicotinic acid (NA) (Figure 1). To maintain NAD+ levels, most NAD+ is recycled via salvage pathways rather than generated de novo. The majority of NAD+ is salvaged from NAM, the product of CD38 and the PARPs (Magni et al., 2004; Magni et al., 1999) or from the various forms of niacin taken up in the diet including NAM, NA, NR and nicotinamide mononucleotide (NMN) (Bogan and Brenner, 2008; Mills et al., 2016; Trammell et al., 2016b; Ummarino et al., 2017). The precursor NR is thought to be directed into the salvage pathway via equilibrative nucleoside transporters (ENTs) (Nikiforov et al., 2011) and converted to NMN by nicotinamide riboside kinases (NRK1/2) (Ratajczak et al., 2016). NR generates unexpectedly high levels of NAAD in mouse liver and heart, as well as in human PBMCs, though the actual mechanism remains to be determined (Trammell et al., 2016a).
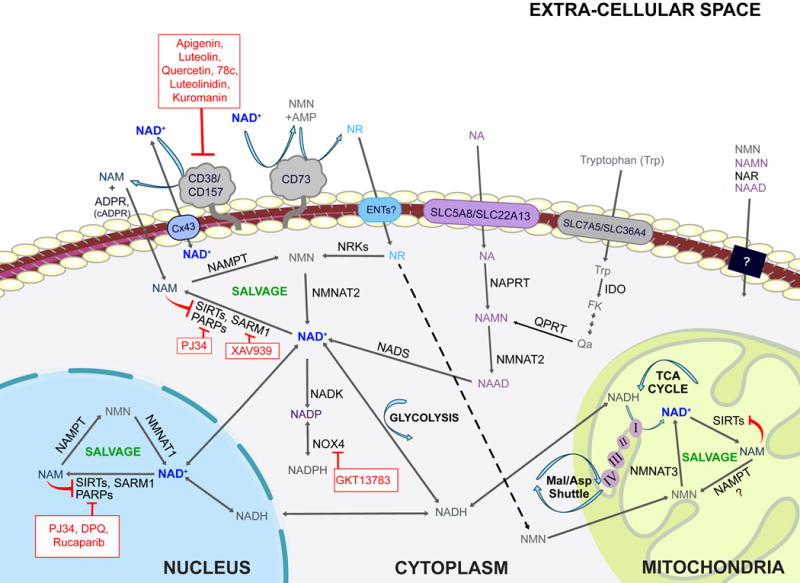
Primary pathways of NAD metabolism
There are two major pathways contributing to NAD synthesis: de novo synthesis and salvage from precursors. The de novo pathway of NAD synthesis converts tryptophan to quinolinic acid (QA) via the kynurenine pathway. The salvage pathways recycle nicotinamide mononuclotide (NMN), nicotinamide riboside (NR), nicotinamide (NAM) and nicotinic acid (NA) in various cellular compartments including the nucleus and mitochondria. These precursors are present in the extracellular milieu and may be transported across the plasma membrane where they are utilized. Extracellular NAD is cleaved by nucleotide phosphatases (CD73) or glycohydrolases (CD38 and CD157). Cleavage by CD73 yields NMN which CD73 can then re-cleave to yield NR. Cleavage by CD38 or CD157 yields NAM. NAM is also produced within cells by NAD+-consuming enzymes such as sirtuins, PARPS and SARM1. NAM and NR are converted to NMN by NAMPT and NRKs respectively. NMN and NAMN then are converted to NAD and NAAD respectively and NAAD is amidated by NADS to yield NAD. Cellular NAD levels may be boosted by activators of the salvage pathway (green) or by inhibitors of enzymes that consume NAD+ such as CD38, PARPs, and SARM1 (red).
The rate of NAD+ synthesis in mammals is largely determined by the first step in the salvage pathway that converts NAM to NMN. In mammals, this is carried out by NAMPT, previously known as visfatin or PBEF (Yang et al., 2007). Levels of NAMPT are highly dynamic, responding to changing cellular demands for NAD+ and cell stresses such as DNA damage and starvation (Yang et al., 2007). NAMPT is an unusual enzyme in that it has both intracellular and extracellular forms known as iNAMPT and eNAMPT (Revollo et al., 2007b). In culture, NAMPT is released into supernatant by various cell types including HepG2 cells, primary hepatocytes, cardiomyocytes and leucocytes (Friebe et al., 2011; Pillai et al., 2013; Schuster et al., 2014) and in vivo, eNAMPT is present in cerebrospinal fluid (Hallschmid et al., 2009) and in serum of both mice and humans (Korner et al., 2007; Revollo et al., 2007a).
The levels of NAMPT expression are believed to control the body’s response to stress, exercise and nutrient status (Frydelund-Larsen et al., 2007; Rappou et al., 2016; Yang et al., 2007) and circadian rhythms (Asher et al., 2008; Masri et al., 2014; Ramsey et al., 2009). Indeed, obesity and high calorie diets reduce both NAMPT (Mercader et al., 2008) and NAD+ levels in various tissues including liver, white adipose (Yoshino et al., 2011), muscle and brown adipose (Canto et al., 2012). Additionally, increased levels of eNAMPT are seen in non-alcoholic fatty liver disease (NAFLD) (Garten et al., 2015), obesity, diabetes mellitus type 2 (T2DM), cardiovascular disease (Chang et al., 2011; Chen et al., 2006; Haider et al., 2006; Saddi-Rosa et al., 2013) and cancer (Audrito et al., 2015). Unlike mammals, invertebrates and yeast convert NAM to NA via Pnc1, a nutrient and stress-responsive NAM deamidase that serves a similar role to NAMPT (Anderson et al., 2003; Revollo et al., 2007a). Some microorganisms are incapable of synthesizing NAD+ de novo and therefore are exclusively reliant on external sources and salvage pathways (Ma et al., 2007).
NAD+ degradation
CD38 and CD157 (BST1) are glycohydrolases that cleave NAD+ to generate NAM and adenosine diphosphoribose (ADPR), while CD38 also hydrolyzes cyclic-ADPR (cADPR) to ADPR. To a lesser extent, both enzymes also act as ADP-ribosyl cyclases that catalyze the hydrolysis of NAD+ to generate NAM and cADPR, a Ca2+-mobilizing second messenger active in many cell types (Berthelier et al., 1998; Kirchberger and Guse, 2013; Malavasi et al., 2008). Both reactions generate NAM, which is rapidly recycled to NAD+ via the salvage pathway. CD38 is ubiquitously expressed while CD157 is expressed primarily in lymphoid tissue and gut (Hernandez-Campo et al., 2006). These enzymes have been implicated in energy metabolism, cell adhesion, various aspects of the immune response and have been linked to human diseases such as Parkinson’s, ovarian cancer, and leukemia (Quarona et al., 2013).
CD38 is a major consumer of NAD+ in mammals. Evidence for this includes the observations that mice lacking CD38 or treated with the CD38 inhibitor apigenin have about 50% more NAD+ (Escande et al., 2013) and that CD38 protein levels increase in various tissues during aging with a concurrent drop in NAD+ levels (Aksoy et al., 2006; Camacho-Pereira et al., 2016). By 32 months of age wildtype mice have about half the NAD+ levels of young mice, whereas CD38 knockout mice maintain their NAD+ levels, and are resistant to the negative effects of a high fat diet (HFD), including liver steatosis and glucose intolerance (Barbosa et al., 2007; Escande et al., 2013). Conversely, mice over-expressing CD38 have lower levels of NAD+, defective mitochondria, decreased oxygen consumption and increased lactate production (Barbosa et al., 2007; Camacho-Pereira et al., 2016). Inhibitors of CD38, such as apigenin and 78c (Haffner et al., 2015) are proof-of-concept molecules for the development of drugs to treat metabolic disorders and possibly other age-related diseases (Chillemi et al., 2013).
SARM1 is a newly discovered NAD+ cleavage enzyme in neurons and possibly other cell types (Conforti et al., 2014; Essuman et al., 2017; Gerdts et al., 2013; Osterloh et al., 2012). It is the first member of a new class of NAD+ consuming enzymes and unique among NADases in that its activity is dependent on SARM1’s Toll/interleukin-1 receptor (TIR)-domain. Moreover, it is the first example of a TIR domain, previously demonstrated to function as a protein interaction domain, possessing enzymatic activity. In response to neuronal injury, the catalytic (TIR)-domain of SARM1 initiates a cell destruction program by converting cytoplasmic NAD+ to ADPR, cADPR and NAM (Essuman et al., 2017). This depletes the cell of NAD+ and triggers axonal degeneration, which can be blocked by overexpression of NAMPT or NMNAT and by supplementation with NR (Gerdts et al., 2015). Knockout of SARM1 rescues the neuronal defects and embryonic lethality of NMNAT2-KO mice that are defective in their ability to salvage NAD+ (Gilley et al., 2015) indicating that SARM1 is a consumer of NAD+ even during embryonic development. Taken together, these results point to SARM1 as an attractive therapeutic target for the treatment of acute neuronal damage and possibly neurodegenerative diseases.
NAD+-responsive signaling pathways
In mammals, the two main NAD+-responsive signaling protein families are the sirtuins and the PARPs (Frye, 1999; Grube and Burkle, 1992; Tanner et al., 2000). Both of these protein families use NAD+ as a co-substrate to modify target proteins, releasing NAM (Bai and Canto, 2012). Sirtuins, first discovered as yeast silencing and telomere protective proteins (Ivy et al., 1985; Rine and Herskowitz, 1987), regulate a wide variety of mammalian proteins involved in processes that include mitochondrial metabolism, inflammation, meiosis, autophagy, circadian rhythms, and apoptosis (Haigis and Sinclair, 2010) (Figure 2). The classic sirtuin reaction is the removal of an acetyl group from lysines on target proteins. The first step is the release of NAM from NAD+, which is followed by the formation of a peptidyl ADP-ribose intermediate covalently attached to the acetyl group of the lysine. The peptide chain with the targeted lysine is then liberated to yield o-acetyl-ADP ribose (OADPR). There are variants on this theme: SIRT5 and SIRT6 have desuccinylation and de-fatty-acylation activities respectively (Du et al., 2011; Jiang et al., 2013), while SIRT4 and SIRT6 can simply liberate NAM, leaving a covalently attached mono-ADP-ribose (Haigis et al., 2006; Liszt et al., 2005).
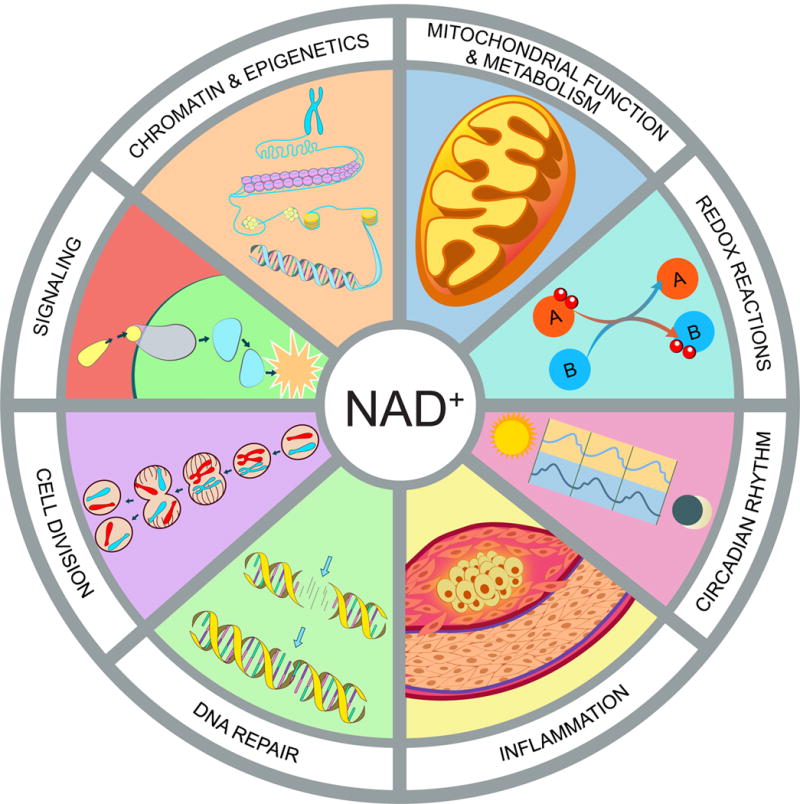
Hallmarks of NAD homeostasis
NAD+ is not merely a redox co-factor, it is also a key signaling molecule that controls cell function and survival in response to environmental changes such as nutrient intake and cellular damage. Fluctuations in NAD impact mitochondrial function and metabolism, redox reactions, circadian rhythm, immune response and inflammation, DNA repair, cell division, protein-protein signaling, chromatin and epigenetics.
The other major NAD+-responsive signaling proteins are the PARPs, which includes both Poly-ADP-ribose polymerases (PARP-1, 2, and 5) and mono-ADP-ribose transferases, which we will refer to as MARTs (PARP-3, 4 6, 10 and 14–16) (Liu and Yu, 2015). PARPs cleave NAD+ and transfer the ADP-ribose moiety to asparagine, aspartic acid, glutamic acid, arginine, lysine and cysteine residues on target proteins, forming branched poly-ADP-ribose polymers and releasing NAM in the process (Bai, 2015). PARP1 and PARP2 are required for numerous cellular processes including DNA repair and transcriptional regulation (Bai et al., 2007; Schreiber et al., 2002; Ziegler and Oei, 2001). PARP1 is best known for hyperactivation in response to DNA damage, depleting the cell of NAD+, most critically in the mitochondria, and thereby inducing apoptosis. The fact that the inhibition of PARP1 raises NAD+ levels in mice, improves mitochondrial function and protects against a HFD (Bai et al., 2011; Pirinen et al., 2014) implies that PARP1 also consumes NAD+ even under physiological conditions.
The functions of other PARPs (PARP3-16) are less well understood. Like PARP1 and 2, PARP3, 5a and 5b have been implicated in DNA-damage repair (Nagy et al., 2016; Rulten et al., 2011). Additionally, PARP5a and 5b, also known as tankyrases, are involved in mitosis and Wnt signaling (Chang et al., 2005; Chi and Lodish, 2000; Huang et al., 2009). PARP16 is a regulator of the unfolded protein response (Jwa and Chang, 2012), and PARP11 is involved in the organization of the nuclear envelope and nuclear pore complexes (Meyer-Ficca et al., 2015). As a component of the vault cytoplasmic ribonucleoprotein, PARP4 is also thought to associate with nuclear pore complexes but its function remains unclear (Chugani et al., 1993; Kickhoefer et al., 1999). PARP7, 10, 12, and 13 are believed to be involved in post-transcriptional regulation of mRNA by RNA-binding domains or ADP-ribosylation of RNA-binding proteins (Bock et al., 2015; Leung et al., 2011). PARP9, 10 and 14 regulate transcription of genes required for immune and inflammatory responses (Iwata et al., 2016; Verheugd et al., 2013). PARP6 regulates hippocampal dendritic development (Huang et al., 2016), and while its function is unknown, PARP15 has been associated with acute myeloid leukemia (Lee et al., 2016). In addition, several PARPs/MARTs may have a role in virus-host interactions (Daugherty et al., 2014).
Due to the role of both sirtuins and PARPs in the response to injury and stress, it is not surprising that there are increasing numbers of examples of the PARPs and sirtuins being co-regulated and regulating each other. For example, the deleted in breast cancer protein 1 (DBC1) binds to and inhibits both SIRT1 and PARP1, the latter of which is dependent on NAD+ binding to the “Nudix Homology Domain” (NHD) of DBC1 (Li et al., 2017). SIRT6 modifies PARP1 by mono-ADP-ribosylation, thereby increasing double strand DNA break (DSB) repair (Mao et al., 2011) whereas inhibition of PARP1 increases expression of SIRT1, SIRT4 and SIRT6 (Wencel et al., 2017).
Pharmacological NAD+ boosters
There are three main approaches that researchers and drug developers are exploring to increase NAD+ levels: supplementation with NAD+ precursors, activation of NAD biosynthetic enzymes, and inhibition of NAD+ degradation (Table 1).
Table 1
Classes of NAD+ boosting molecules and known effects in humans
| Mechanism of Action | Pharmacological Agent | Health Outcomes Observ | References |
|---|---|---|---|
| NAD+ Precursors | Niacin (NA) | ↓ TC and LDL in dyslipidemia ↓ Serum phosphate after kidney injury ↑ GFR after kidney injury ↓ Risk of myocardial infarct ↓ Risk of stroke ↓ Risk of CVD mortality ↑ Recovery from schizophrenia ↓ Rate of cognitive decline in AD ↓ Rate of metastases/relapse in breast cancer ↑ HDL-C in sickle cell disease |
(Abram Hoffer, 2008; Garg et al., 2017; Morris et al., 2004; Phelan et al., 2017; Premkumar et al., 2007; Rennie et al., 2015; Scoffone et al., 2013) |
| NR | ↑ NAD+ in blood (dose-dependent) No Adverse effects |
(Trammell et al., 2016a) | |
| NAM | |||
| NaR | |||
| NaMN | |||
| NAAD | |||
| NMN | |||
| IHN | |||
| CD38 Inhibitors | Quercetin | ↓ LDL in CVD ↓ Blood pressure ↓ BMI ↓ Waist circumference ↓ Triglycerides |
(Dower et al., 2015; Katske et al., 2001; Lee et al., 2016a; Menezes et al., 2017; Pfeuffer et al., 2013) |
| Luteolin | ↓ Serum levels of IL-6 and TNF ↑ Adaptive functioning in autism |
(Tsilioni et al., 2015) | |
| Apigenin | |||
| 78c | |||
| Luteolinidin | |||
| Kuromanin | |||
| PARP Inhibitors | BGB-290, Olaparib, Rucaparib, Veliparib, CEP-9722, E7016, Talazoparib, Iniparib | ↓ Tumor size, metastases, and relapse in breast and ovarian cancer | (Brown et al., 2016; Evans and Matulonis, 2017) |
| Niraparib (MK – 4827) | |||
| PJ34 | |||
| DPQ | |||
| 3-aminobenzamide | |||
| SARM Inhibitors | XAV939 | ||
| NAMPT Activators | P7C3 |
NAD precursors
Homeostatic levels of NAD+ can be achieved by ingesting 15 mg of niacin daily. For most of the 20th century, this was considered optimal. It is now known that NAD+ levels decline with age and that raising levels back up to, or even above baseline, provides a surprising number of health benefits in a wide range or organisms, from yeast to rodents. The first evidence that NAD-boosting could increase lifespan came from studies our laboratory performed in yeast cells over 15 years ago. The yeast gene PNC1, encoding the enzyme that catalyzes the first step in the NAD+ salvage pathway from nicotinamide, is one of the most highly upregulated yeast genes in response to environmental stresses including heat, osmotic stress, and the restriction of amino acids and glucose (CR). We reasoned that the ability of these mild stresses to extend lifespan might be due to the upregulation of the PNC1 gene. Indeed, the constitutive overexpression of PNC1 was sufficient to increase the stress resistance and lifespan of yeast cells, whereas deletion of PNC1 rendered them unable to respond positively to caloric restriction and increased temperature (Anderson et al., 2003). In Drosophila, overexpression of the Pnc1 homolog D-NAAM extends mean lifespan by 30% (Balan et al., 2008), indicating that the upregulation of NAD synthesis might be a conserved longevity mechanism.
It has been challenging to constitutively upregulate NAD+ levels in mammals for reasons that are unclear (Frederick et al., 2015). The most common and effective approach has been a pharmacological one. NR and NMN are soluble and orally bioavailable endogenous molecules, making them the molecules of choice for animal experiments and human clinical trials (Conze et al., 2016; Trammell et al., 2016a). In rodents, NR is more efficient in boosting NAD+ than NA and NAM (Trammell et al., 2016a), possibly due to increased uptake (Imai, 2009; Imai and Guarente, 2014; Ratajczak et al., 2016; Revollo et al., 2007a).
The ability of NA to reduce cholesterol levels has been known for some time (Altschul et al., 1955) but only recently has this effect been ascribed to its ability to boost NAD+ levels (Canto et al., 2012). Though several studies have shown that NA can raise NAD+ levels (Canto et al., 2012; Kaneko et al., 2006; Santidrian et al., 2013), its use has been limited by unpleasant side effects that include flushing and itching of the skin caused by prostaglandin release (Birjmohun et al., 2005; Chen and Damian, 2014; Rolfe, 2014) and the fact that NAM inhibits PARPs and sirtuins (Bitterman et al., 2002).
Activation of NAD+ synthesis
An alternative approach to boost NAD+ levels is to directly activate NAD+ biosynthetic enzymes, in particular those that catalyze the rate-limiting steps of de novo synthesis and salvage pathways. Several enzymes are currently under investigation. The NAM salvage pathway is the predominant route in mammalian NAD+ biosynthesis (Magni et al., 1999) and NAMPT is the rate-limiting enzyme in the conversion of NAM to NAD+ (Revollo et al., 2007a). Under normal conditions NAMPT activity is sustained to maintain NAD+ homeostasis. NAMPT activity, however, declines with age and is exacerbated by acute lung injury, atherosclerosis, cancer, diabetes, rheumatoid arthritis and sepsis. Increasing systemic NAD+ biosynthesis with small chemical NAMPT activators is considered an attractive therapeutic approach. Several NAMPT activating compounds were apparently identified using a fluorometric NAMPT activity assay, but in that study only the inhibitors were characterized (Zhang et al., 2011). A neuroprotective agent, P7C3, may be a relatively weak NAMPT activator in vitro (Wang et al., 2014) although this result has not yet been replicated. NMNATs are also attractive targets for raising NAD+ in cells because they have dual substrate specificity for NMN and NaMN and contribute to both de novo and salvage pathways (Zhou et al., 2002). The green tea compound epigallocatechin gallate (EGCG) has been reported to activate NMNAT2 by more than 100% and NMNAT3 by 42% at 50 μM, although this needs to be confirmed, as no data was presented in the paper (Berger et al., 2005).
Inhibition of NAD+ degradation
An alternative approach to raising NAD+ is to inhibit its degradation either by inhibiting PARPs or NADases, also known as glycohydrolases. The major NADase in mammals, CD38, is inhibited in vitro at low micromolar concentrations by flavonoids including luteolinidin, kuromanin, luteolin, quercetin and apigenin (IC50 <10 μM) (Escande et al., 2013; Kellenberger et al., 2011). These molecules also appear to target CD38 in vivo. Apigenin increases NAD+ levels in multiple tissues, decreases global proteome acetylation and improves glucose and lipid homeostasis in obese mice, ostensibly by increasing activity of SIRT1 and SIRT3 (Camacho-Pereira et al., 2016; Escande et al., 2013). Similarly, luteolinidin prevents the loss of NAD+ and preserves endothelial and myocardial function in the post-ischemic heart (Boslett et al., 2017). GlaxoSmithKline developed thiazoloquin(az)olinones such as the compound “78c”, which have greater potency than the flavonoids and can boost NAD+ levels in plasma, liver and muscle (Haffner et al., 2015). The target specificity of 78c and whether or not it enters cells rather than acting on extracellular CD38 remain unknown. PARP1 inhibitors are marketed as adjuncts to chemotherapy or monotherapies for cancer (Brown et al., 2016). SARM1 is another NADase, which initiates a local axonal degeneration program after nerve injury that involves the rapid breakdown of NAD+ to ADPR, cADPR and NAM (Essuman et al., 2017; Gerdts et al., 2015; Summers et al., 2016). XAV939, a putative SARM1 inhibitor that also inhibits PARP5a (TNKS) and 5b (TNKS2), was identified in a chemical genetic screen nearly a decade ago (Huang et al., 2009), though its potential to boost NAD+ has only recently become evident (Gerdts et al., 2015). XAV939 has excellent pharmacokinetic properties and is currently in clinical development to treat neurological disorders and axonal injury (Haikarainen et al., 2014). To date, it is unclear if XAV939 functions in vivo by inhibiting SARM1 or other targets.
Uptake and biodistribution of NAD+ precursors
How NAD+ and its precursors are taken up by cells and tissues is not well understood and is currently the subject of considerable research and some debate. It is generally agreed that tryptophan is taken up by SLC7A5 and SLC36A4 (Kanai et al., 1998) whereas NA is taken up by SLC5A8 and SLC22A13 (Bahn et al., 2008; Gopal et al., 2007) (see Figure 1). NAM, an uncharged molecule, rapidly diffuses across the plasma and mitochondrial membranes, consistent with the fact that NA and NAM have additive effects on raising NAD+ in cells (Hara et al., 2007). For larger charged molecules, specific transporters must exist. In general, cells are unable to take up NAD+, the one exception being neurons (Araki et al., 2004). How neurons take up NAD+ is not known. Bacteria transport NAD+ via ATP/ADP translocase (Haferkamp et al., 2004), though a mammalian equivalent NAD+ transporter has yet to be identified. In yeast, NR and NAR are transported by Nrt1 and an equilibrative nucleoside transporter (ENTs), FUN26 (Belenky et al., 2008). The same may be true in mammals based on a reduction in cellular NAD+ levels when the nucleoside transporter is inhibited by dipyridamole (Nikiforov et al., 2011), but there is no direct evidence of a specific transporter.
NAD+ is present in serum and the extracellular space and some data suggests that this is also the case for NMN (Revollo et al., 2007b). These data imply that circulating NAD+ precursors might be acting to coordinate NAD+ biosynthesis and mediate signals between organs. Indeed, the existence of the NMN generating enzyme eNAMPT in serum fits with this model, though some argue that NMN is not detectable in the extracellular milieu and that conditions do not favor catalytic activity (Audrito et al., 2015; Hara et al., 2011; Ratajczak et al., 2016; Revollo et al., 2007a; Revollo et al., 2007b). Under physiological conditions, the concentrations of NAD+ and NAM in extracellular fluids such as blood plasma are in the micromolar range and can be boosted by oral administration of NMN or NR (Trammell et al., 2016a).
Extracellularly, NAD+ is cleaved to NMN and AMP by nucleotide pyrophosphatases (NPPs) including CD73 (Aleo et al., 2001; Grozio et al., 2013) while the transmembrane glycohydrolases CD38 and CD157 cleave NAD+ to generate NAM and adenosine diphosphoribose (ADPR) and can also act as ADP-ribosyl cyclases that catalyze the hydrolysis of NAD+ to generate NAM, and cADPR, a cell-cell messenger that appears to mediate activated monocyte-induced Ca2+ signaling, ROS production, and apoptosis (Yang et al., 2011).
Whether or not NMN is taken up by a transporter is currently the subject of debate (Mills et al., 2016; Ratajczak et al., 2016). Brenner, Canto and colleagues argue that NMN is not taken up fast enough to invoke the presence of a transporter, and that both NAD+ and NMN undergo extracellular degradation to generate permeable precursors that can be taken up by cells (Ratajczak et al., 2016). On the other hand, Imai argues that this is likely a cell-type specific phenomenon and that some cell types can rapidly take up NMN (Mills et al., 2016). If so, the identification of the putative transporter will help resolve the debate and help identify which cell types and tissues are able to transport NMN across the plasma membrane. Additional studies with isotopically labeled NAD+ precursors to trace the uptake and metabolism of these molecules should help answer these questions.
The metabolism and biodistribution of NAD precursors in various tissues and within cells is poorly understood. The system is clearly complex, arising from variation in transporter expression, enzymatic machinery, extracellular degradation and resynthesis (Bogan and Brenner, 2008; Duarte-Pereira et al., 2016; Ratajczak et al., 2016; Zamporlini et al., 2014). NAMPT is expressed ubiquitously in the body but there are large differences in the levels of expression between tissues (Dietrich et al., 1966; Revollo et al., 2007b). Metabolic profiling of mouse tissues indicates that the activity of NMNAT isoforms, required for amidated NAD+ salvage pathways, which utilize NR or NAM, is much higher than NAMPT and is non-rate-limiting in most tissues, except blood. NADS activity, required for the deamidated pathway that utilizes NA, appears to be rate limiting in lung and skeletal muscle where levels are difficult to detect and the enzyme’s substrate, NAAD, accumulates (Mori et al., 2014). In brain and heart, the NAMPT-dependent pathway is the preferred route of salvage, whereas in skeletal muscle, NRK-dependent salvage pathways are the preferred mode for generating NAD+. Additionally, expression analysis of NRK subtypes shows that NRK1 is expressed ubiquitously while NRK2 is mainly present in skeletal muscle (Fletcher et al., 2017). Consistent with this, chronic NR supplementation raises NAD+ levels in muscle, but not in brain or white adipose tissue (Canto et al., 2012). Interestingly, the CD38 inhibitor 78c increases NAD+ by more than 5-fold in mouse liver but only >1.2-fold in muscle, arguing that CD38 activities are also tissue specific (Haffner et al., 2015). Whether or not the ability of CD38 to raise NAD+ levels in tissues is due to intra- or extracellular activity remains to be determined (Bonkowski and Sinclair, 2016; Gupte et al., 2017).
Effects of NAD+ boosters on physiology and health in mouse models
The initial discovery that genetically upregulating NAD+ biosynthesis can increase the stress resistance and lifespan of yeast cells and Drosophila (Anderson et al., 2002; Anderson et al., 2003; Balan et al., 2008), prompted investigation of NAD+ boosters in rodents, both wildtype and disease models, often with dramatic effects (Figure 3).
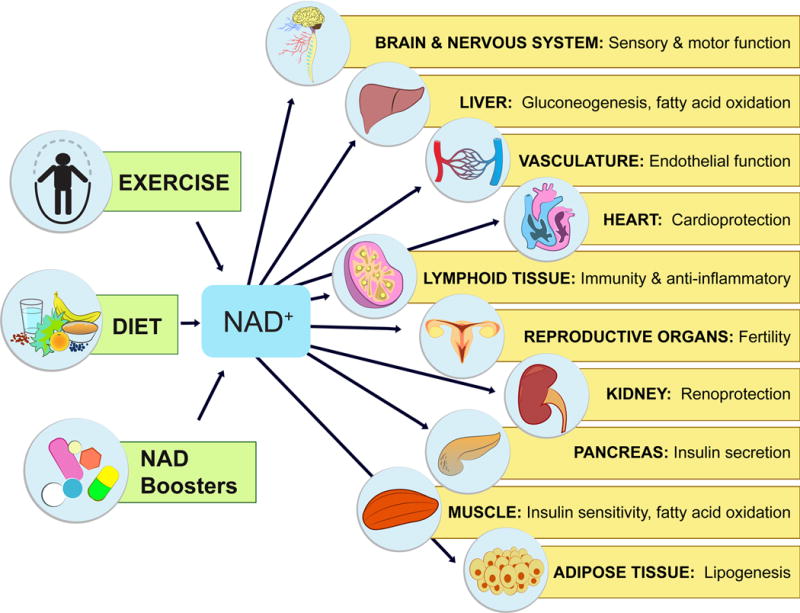
Physiological effects of NAD-boosting molecules
NAD+ levels steadily fall as we age, leading to a decline in the function of cells and organs. By raising NAD+, NAD+ boosters can have profound effects on the health and survival of mammals. Increases in NAD+ promote cognitive and sensory function, gluconeogenesis in liver, lipogenesis in adipose tissue, insulin secretion in pancreas, and insulin sensitivity in muscle. NAD+ also promotes endothelial cell proliferation and protects against cardio- and cerebrovascular disease. NAD regulates immune function and inflammation and, protects against acute injury in kidney. NAD promotes and extends fertility in both males and females, ostensibly by activation of sirtuins.
Liver Function
Key enzymes in NAD+ signaling pathways are known to protect the liver from fat accumulation, fibrosis and insulin resistance, which are related to the development of fatty liver diseases, such as NAFLD and NASH. SIRT1 and its downstream targets PGC-1α, PSK9, and SREBP1 maintain mitochondrial, cholesterol transport, and fatty acid homeostasis (Baur et al., 2006; Picard et al., 2004; Rodgers et al., 2005; Yang et al., 2006). SIRT2 controls gluconeogenesis by deacetylating phosphoenolpyruvate carboxykinase (PEPCK) (Jiang et al., 2011), SIRT3 regulates OXPHOS (Ahn et al., 2008), fatty acid oxidation (Hirschey et al., 2010), ketogenesis (Shimazu et al., 2010), and defense against oxidative stress (Finley et al., 2011; Someya et al., 2010), while SIRT6 controls gluconeogenesis (Dominy et al., 2012; Zhang et al., 2014).
Given the critical nature of these pathways in the liver, the maintenance of NAD+ levels is imperative for optimal organ function. As a result of obesity and aging, levels of NAMPT decline and CD38 levels increase, leading to a 2-fold decrease in steady state NAD+ levels by mid-age (Zhou et al., 2016). Raising NAD+ levels back to those of young or lean mice has been particularly effective at preventing and treating obesity, alcoholic steatohepatitis and NASH, while improving glucose homeostasis and mitochondrial dysfunction. Approaches that have worked well in rodents include the inhibition of NAD+ consuming enzymes such as PARPs (Bai et al., 2011; Gariani et al., 2017) and CD38 (Barbosa et al., 2007), the inhibition of nicotinamide N-methyltransferase (Nnmt) (Kraus et al., 2014) and supplementation with NAD+ precursors, NR (Canto et al., 2012) or NMN (Yoshino et al., 2011). NAD boosting appears to not only improve the health of the liver, but also increase its capacity for regeneration and protect it against hepatotoxicity. After partial hepatectomy (PHx), NR-treated mice have increased and more uniform liver regeneration, a shorter period of steatosis, increased DNA synthesis, and improved lipid metabolism as compared to untreated control mice (Mukherjee et al., 2017). Olaparib treatment raises NAD+ levels and prevents lipopolysaccharide (LPS)-induced acute hepatitis (Gariani et al., 2017). NAM and another PARP inhibitor, PJ34, are both effective in increasing NAD+ levels, and preventing hepatosteatosis and thymus atrophy, in a chick embryo model of dioxin-toxicity (Diani-Moore et al., 2017).
Kidney Function
Several lines of evidence indicate that reduced levels of NAD+ in aged kidneys and a corresponding decrease in sirtuin activity are largely responsible for reduced kidney function and resilience with age (Ugur et al., 2015). Consistent with this, activation of SIRT1 and SIRT3 by NAD+ supplementation protects against high-glucose-induced kidney mesangial cell hypertrophy (Zhuo et al., 2011) while treatment of mice with NMN protects from cisplatin-induced acute kidney injury (AKI) in a SIRT1-dependent manner (Guan et al., 2017). 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR), a nucleoside converted to 5-amino-4-imidazolecarboxamide riboside 5′-monophosphate (ZMP) that stimulates AMPK activity, increases NAD+ levels and also protects against cisplatin-induced AKI in a SIRT3-dependent manner (Morigi et al., 2015). NAM supplementation in mice stimulates secretion of the kidney-protective prostaglandin PGE2, improves renal function after ischemia and similarly prevents cisplatin-induced AKI, ostensibly by stimulating NAD+ synthesis (Tran et al., 2016).
Skeletal Muscle Function
Old mice have increased markers of muscle atrophy and inflammation, as well as impaired insulin signaling and insulin-stimulated glucose uptake compared to young wild-type mice. Treatment of old mice with NAD+ precursors, such as NR and NMN, dramatically improves muscle function. For example, treatment of old mice with NMN (500 mg/kg/d i.p. for 7 days) reverses detrimental age-associated changes in muscle, by increasing mitochondrial function, increasing ATP production, reducing inflammation, and switching glycolytic type II muscle to a more oxidative fiber type (Gomes et al., 2013). Similarly, old and high fat diet (HFD)-fed mice treated with NR (400 mg/kg/d in chow) have improved oxidative metabolism in muscle as well as increased endurance and thermogenic capacity (Canto et al., 2012). NR also increases the number and quality of muscle stem cells (MuSC) and enhances muscle regeneration in old mice, in part, by improving mitochondrial function and preventing MuSC senescence (Zhang et al., 2016). In the context of muscular dystrophy models, NR improves mobility in dystrophic C. elegans mutants (Ryu et al., 2016), NAD+ supplementation of zebrafish reduces the percentage of detached muscle fibers and increases mobility (Goody et al., 2012), and in the MDX mouse, NR improves mitochondrial function, recovery from muscle injury, and running capacity (Ryu et al., 2016).
Cardiac Function
NAD+ levels are critical for normal heart function and recovery from injury. Of all the NAD+−dependent signaling proteins, SIRT3 seems to be the most important in this context. SIRT3 knockout mice have hyperacetylated OXPHOS enzymes, reduced ATP (Sundaresan et al., 2009) and are hypersensitive to aortic constriction, ostensibly due to activation of CypD, a regulator of the mitochondrial permeability transition pore (Hafner et al., 2010; Sundaresan et al., 2009). These mice develop fibrosis and cardiac hypertrophy as early as 13 months of age, which is further exacerbated by the loss of SIRT3 in aged and hypertrophic hearts, a decline that can be reversed by treatment with NMN (Horton et al., 2016). Consistent with this, NAMPT overexpression and NMN treatment either 30 min before ischemia (500 mg/kg, i.p.) or repetitive administration just before and during reperfusion, provides marked protection against pressure over-load and ischemia/reperfusion (I/R) injury, reducing infarct size by as much as 44% (Hsu et al., 2009; Karamanlidis et al., 2013; Pillai et al., 2005; Yamamoto et al., 2014). Treatment with NAD+ precursors also improves heart function in old MDX mice with cardiomyopathy (Ryu et al., 2016), improves mitochondrial and cardiac function in a mouse model for iron deficiency-induced heart failure (Xu et al., 2015) and restores cardiac function to near normal levels in a mouse model of Friedreich’s Ataxia (FRDA) cardiomyopathy (Chan et al., 2013; Martin et al., 2017).
Endothelial and Vascular Function
Cardiovascular and cerebrovascular diseases contribute to the greatest decline in quality of life after 65 and are directly responsible for about one third of all deaths (Nichols et al., 2014; Ungvari et al., 2010). Understanding why this occurs and how to reverse it is key to future gains in human health. Treatment of old mice with NMN (~300mg/kg/d for 8 weeks) restores carotid artery endothelium-dependent dilation (EDD), a measure of endothelial function, while reducing aortic pulse wave velocity and elastic artery stiffness (de Picciotto et al., 2016). The performance of most organs and tissues is critically dependent on an abundant, fully functional microcapillary network that maintains a supply of oxygen, exchanges heat and various nutrients, and removes the waste products of metabolism. Yet one of the most profound changes to the body as it ages is a decline in the number and function of endothelial cells (ECs) that line the vasculature. Treatment of mice with NMN (500 mg/kg/d in water for 28 days) improves blood flow and increases endurance in elderly mice by promoting SIRT1-dependent increases in capillary density (Das et al., 2018 Das et al., in press at Cell, MS included in submission). Thus repleting NAD+ levels in the vascular endothelium is an attractive approach to increasing mobility in the elderly and treating conditions exacerbated by decreased blood flow, such as ischemia reperfusion injury, slow wound healing, liver dysfunction, and muscle myopathies.
DNA Repair and Cancer
Because NAD+ is involved in so many aspects of cancer biology, from mitochondrial activity to cell survival, there are a variety of ways it could be used in the clinic. The rationale for reducing NAD+ levels in tumors is that they will be less able to repair DNA damage, thereby increasing their sensitivity to chemotherapeutic agents. Natural inhibitors of PARP1 include the flavonoids myricetin, tricetin, quercetin, fisetin, rutin and naringin (Geraets et al., 2007). More potent PARP1 inhibitors, such as PJ34, DPQ, and the marketed drugs olaparib and rucaparib sensitize tumors to DNA damage, although the drugs have limited use because of toxicity (Alano et al., 2010; Almeida et al., 2017; Mathews and Berk, 2008). Inhibitors of NAMPT, such as FK866 and KPT-9274, selectively kills cancer cells, and are currently being tested for efficacy in patients with solid malignancies (Chini et al., 2014; Poljsak, 2016; Sharif et al., 2016). Inhibition of indoleamine 2,3-dioxygenase (IDO), which mediates the first step in the conversion of tryptophan in de novo NAD+ synthesis, shows promise in mice as an antitumor therapeutic target (Friberg et al., 2002; Muller et al., 2005; Uyttenhove et al., 2003).
The other approach to treating cancer has been to increase NAD+ levels, the rationale being that an excess of NAD+ will boost mitochondrial respiration and downregulate glycolysis, counteracting Warburg metabolism that cancer cells prefer (Wu et al., 2014). Increased NAD+ would also boost the activity of SIRT1 and SIRT6, both of which can inhibit tumors by downregulating beta-catenin signaling and glycolysis (Firestein et al., 2008; Sebastian et al., 2012). One concern is that raising NAD+ may promote DNA repair and angiogenesis, helping cancer cells thrive (Batra and Kislay, 2013; Fang et al., 2016; Li et al., 2017). Long-term studies of wildtype mice, however, failed to provide any evidence of increased tumor size or number (Shukla et al., 2014; Tummala et al., 2014). Interestingly, overexpression of NMNAT3 raises mitochondrial NAD+ and inhibits the growth of glioblastoma cells (Son et al., 2017) and supplementation with NA or NAM inhibits tumor growth and multi-organ metastasis in SCID mice (Santidrian et al., 2013). It will be important to test the effects of NAD+ boosters in gold-standard cancer models, either in the absence of, or combination with, standard chemotherapeutic agents.
Immunity and Inflammation
There is a growing body of evidence that NAD+ precursors can have anti-inflammatory effects. Treatment of 24-month-old mice with NMN for one week reduced the expression of inflammation markers such as TNFα and IL-6 in skeletal muscle (Gomes et al., 2013). Similarly, NR significantly reduced inflammation in a mouse model of AT autoimmunity (Fang et al., 2016) and in the muscular dystrophy MDX mouse model, though it is not clear if these effects are secondary to an improvement in muscle function (Ryu et al., 2016). NAM has been effective in the treatment of various inflammatory skin conditions (Niren, 2006), reduces the area of infiltration and demyelination in experimental autoimmune encephalomyelitis (EAE) mouse models (Kaneko et al., 2006) and prevents photo-immunosuppression and photo-carcinogenesis (Damian et al., 2008; Gensler, 1997; Yiasemides et al., 2009). However, it is not yet known if these effects are due to increaseing NAD+ levels or other physiological effects.
NAD+ metabolism also plays an important role in host-pathogen interactions. Several pathogenic organisms are auxotrophic and therefore dependent on the host for NAD+ or precursors. Targeting the uptake of these precursors in pathogens may be a fruitful avenue to pursue (Domergue et al., 2005; Ma et al., 2007; Moreira et al., 2015; Murray et al., 1995; Zerez et al., 1990). Other organisms have their own NAD+ salvage pathways that can be targeted by drugs to kill the pathogen. In fact, the antibiotic pyrazinamide, commonly used to treat tuberculosis, is converted to a toxic product by a pyrazinamide/nicotinamidase called PncA, which catalyzes the first step in the conversion of NAM back to NAD+. As with cancer, the biology of the host-pathogen interaction is complex and has produced conflicting results, depending on which downstream player, whether it be PARP1, CD38 or SIRT1, is being analyzed (Koedel et al., 2002; Moreira et al., 2015; Partida-Sanchez et al., 2007; Ren et al., 2014). Further investigation will be required to determine if NAD+ depletion or repletion in the host can be used as treatment for infections.
Neuronal Function
The neuroprotective effects of NAD+ precursors were first revealed by a study of middle cerebral artery (MCAO)-induced ischemia, where treatment with NAM reduced the extent of infarct in Wistar rats (Ayoub et al., 1999; Klaidman et al., 2003; Sadanaga-Akiyoshi et al., 2003), a finding recently replicated by treatment with NMN (Wei et al., 2017a). Though these early findings were indicative, it wasn’t until 2004 that NAD+ was brought into the forefront of neuroscience (Gerdts et al., 2015; Sasaki et al., 2016). The Wallerian degeneration slow (Wlds) mouse is so named for its neurons that are relatively resistant to axonal degeneration after nerve damage. The mutation was known to be due to an autosomal dominant mutation that fused the ubiquitination factor e4b (Ube4b) with NMNAT1 (Coleman et al., 1998), but the role of NAD+ was not appreciated until it was shown that increased NAD+ could mimic the Wlds mutation, ostensibly by activating SIRT1 (Araki et al., 2004). The same group recently identified neuronal SARM1 as the major cause of NAD+ depletion in injured neurons (Essuman et al., 2017).
Since then, numerous studies have reinforced the view that NAD+ levels are key to neuronal function and survival. This includes the dependence on NMNAT2 and its NAD synthesis activity for axonal survival (Yan et al., 2010). There is some evidence indicating that NMN treatment promotes axon degeneration, and that the dependence on NMNAT2 for axon survival is due to its consumption of NMN rather than its NAD synthesis activity (Di Stefano et al., 2015). This is supported by recent in vivo work in zebrafish larvae and mice showing that expression of a bacterial NMN-deamidase, which consumes NAD but does not have NAD synthesis activity, rescues axonal defects and promotes axon survival (Di Stefano et al., 2017). On the other hand, NMN preserves hippocampus-dependent spatial memory after forebrain ischemia (Park et al., 2016) and reduces edema, oxidative stress, inflammation, and neuronal death in a mouse collagenase-induced intracerebral hemorrhage (cICH) model (Wei et al., 2017b).
In addition to protecting damaged neurons, NAD+ precursors have shown promise in delaying the effects of several neurodegenerative diseases. In models of Alzheimer’s Disease (AD), NR and NMN treatment improve cognition and synaptic plasticity in mice and rats (Gong et al., 2013; Hou et al., 2018; Long et al., 2015; Sorrentino et al., 2017; Wang et al., 2016). NAM increases cell viability in a Drosophila model of PD (Jia et al., 2008) and several studies have also suggested that an NA-rich diet both reduces the risk of developing Parkinson’s Disease (PD) and improves the physical functioning of individuals with PD (Alisky, 2005; Fall et al., 1999; Hellenbrand et al., 1996), who typically have low kynurenine/tryptophan ratios (Widner et al., 2002). Both NR and NMN improve motor function and memory in worm and mouse models of ataxia telangiectasia (AT) (Fang et al., 2016). Furthermore, P7C3, a pro-neurogenic putative NAMPT activator (Pieper et al., 2010; Wang et al., 2014), is neuroprotective in animal models of Parkinson’s Disease and ALS (De Jesus-Cortes et al., 2012; Tesla et al., 2012). NAD-boosting regimens prevent and in some cases can reverse neuronal degeneration associated with hearing loss, prion toxicity, retinal damage, traumatic brain injury (TBI), and peripheral neuropathy (Brown et al., 2014; Dutca et al., 2014; Hamity et al., 2017; Lin et al., 2016; Vaur et al., 2017; Yin et al., 2014; Zhou et al., 2015).
Aging and Longevity
Total NAD+ levels were once considered extremely stable. Recently, however, it has become clear that a steady decline in total NAD+ levels over time is a natural part of life for all species, from yeast to humans (Balan et al., 2008; Belenky et al., 2007; Lin et al., 2004; Massudi et al., 2012; Mouchiroud et al., 2013; Zhang et al., 2016; Zhu et al., 2015). This decline, along with the decreased activity of NAD+ signaling proteins, is believed to be one of the major reasons organisms, including humans, age.
Given that budding yeast were first shown to live longer when the salvage pathway was upregulated, it was only appropriate that NAD+ precursors were first shown to extend lifespan in this species. Yeast grown on 10 μM NR have increased gene silencing, decreased rDNA recombination (a known cause of aging in yeast), and an increased lifespan of >4 generations (Belenky et al., 2007). Both NR and NMN improve neuronal and muscular function and extend the lifespan of C. elegans by >10% (Mouchiroud et al., 2013).
In mice, raising NAD+ levels has been effective in delaying progeroid (accelerated aging) phenotypes and extending lifespan in various models including BubR1 hypomorphs, xeroderma pigmentosum (XPA), ataxia telangiectasia (AT), and a Cockayne’s syndrome mouse model (Fang et al., 2016; Fang et al., 2014; North et al., 2014). In the case of the BubR1 mouse, median lifespan was extended by 58% percent and maximum by 21% (North et al., 2014), which is more than twice the effect of senolytics in this model (Baker et al., 2011). In part because of the time and cost, the number of studies testing NAD+ boosters in mouse lifespan are few. Treatment of old (20-month-old) mice with NR extended their lifespan by nearly 5%, even when started at an age where few treatments work well, with the exception of rapamycin. Increased lifespan was also associated with a variety of physiological benefits including improved mitochondrial function and preservation of stem cell function (Zhang et al., 2016). Though NMN or other NAD+ boosters have not yet been tested for their effects on murine lifespan, some mice have been dosed for long periods. For example, starting at 5 months, NMN was administered to mice for over a year. Treated mice had increased activity, improved insulin sensitivity and lipid profiles, improved vision, and greater bone-density (Mills et al., 2016). Taken together, these results along with the various health benefits and age-reversal activities listed above, support the possibility of using NAD+ boosters as therapeutics against a broad range of age-associated diseases and possibly as a way to delay aging and age-related physical decline.
Human trials
NAD+ boosters have shown efficacy in a variety of mouse models of human disease (Figure 4), prompting numerous clinical trials of NAD+ boosters in humans (Table 1). The most studied of the NAD+ precursors in humans is NA (niacin). In large doses greater that a gram, NA is an effective way to treat hypercholesterolemia as it lowers LDL,and is one of the few drugs that significantly raises HDL (Altschul et al., 1955; Garg et al., 2017; Rivin, 1962). It is commercially available either as compressed NA (Niacor®) or extended-release to prevent flushing caused by prostaglandin release (Niaspan®, Advicor®, Simcor®). Niaspan® was first approved for several indications by the FDA in 1997 and later used in combination with statins for the treatment of primary hyperlipidemia and mixed dyslipidemia. NA and NAM have been or are currently being evaluated for other health benefits including treatments for acne, kidney diseases, lupus, Alzheimer’s disease, schizophrenia, Diabetes Mellitus, non-small-cell lung carcinoma, obesity, HIV induced dyslipidemia, non-alcoholic fatty liver disease (NAFLD), sickle cell disease and others. Though NA has been shown to raise NAD+ levels in rodents (Canto et al., 2012; Santidrian et al., 2013), the possibility that NA improves cholesterol profiles in humans, at least in part, by raising NAD+ levels (Gariani et al., 2016) is generally not discussed in the literature.
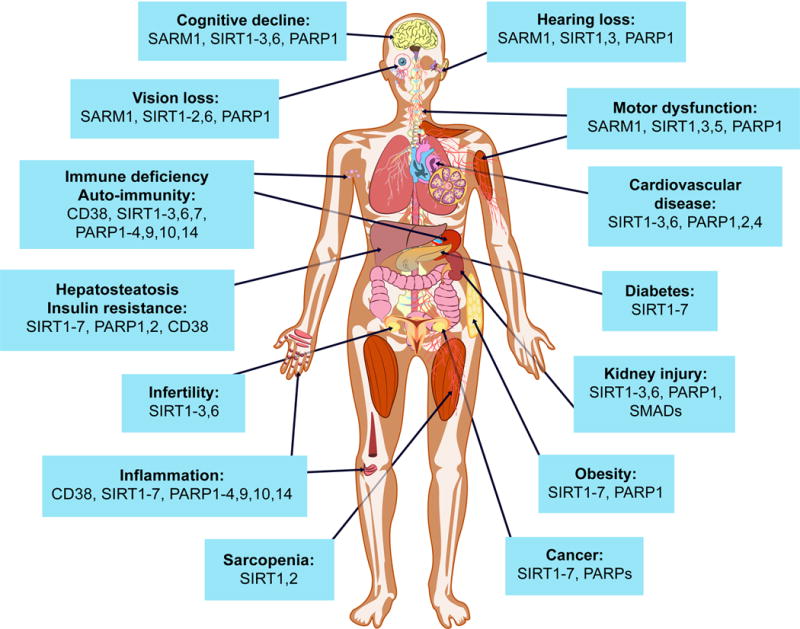
Potential impact of NAD+ boosters on human health via NAD+ signaling pathways
A decline in NAD+ during aging is believed to be a major cause of disease and disability, such as hearing and vision loss, as well as cognitive and motor dysfunction, immune deficiencies, auto-immunity and dysregulation of the inflammatory response leading to arthritis, metabolic dysfunction and cardiovascular disease. In mouse models, NAD+ boosters prevent or treat a variety of different diseases, prompting a search for NAD+ boosters that are safe and effective as drugs to treat both rare and common diseases, and potentially aging itself.
In rodents, NAD+ boosters such as NR and NMN appear to raise NAD+ to greater levels than NA and NAM, though no head-to-head study has yet been performed. NR is currently sold as a supplement either alone or in combination with methylated resveratrol by companies such as Elysium Health and HPN that typically contain “NIAGEN®”. Research into the effects of NR and NMN in humans is gaining considerable traction, with a number of registered clinical trials being recently completed or currently underway (see Clinicaltrials.gov). For example, a randomized, double-blind, three-arm crossover pharmacokinetic study in 12 human subjects showed that NR raises NAD+ by as much as 2.7-fold in human blood with a single oral dose of 1000 mg, with NAAD emerging as a sensitive biomarker (Trammell et al., 2016a). Researchers at the University of Washington completed a clinical trial with 140 participants showing that orally administered NR gives a dose dependent increase in NAD+ from 250–1000 mg/d plateauing at a 2-fold increase in NAD+ at day 9 (Airhart et al., 2017). Another study reported positive effects of NR on vascular endothelial function in healthy middle-aged and older adults, with further investigations of motor and cognitive changes to come (Heilbronn, 2017). At least seven other studies are now underway assessing the effects of NR on such parameters as muscle mitochondrial function, cognition, immune function, kidney function, traumatic brain injury, brown fat activity, lipid accumulation, energy metabolism, cardiovascular risk, body composition, and acetylcarnitine levels.
An international collaborative team between Keio University in Tokyo and Washington University School of Medicine in St Louis is running a Phase I human clinical study of NMN in Japan (Tsubota, 2016). Clinical trials examining the safety and efficacy of NMN are also currently being run at Washington University, investigating the effect on insulin senstivity, endothelial function, lipids, body and liver fat and markers of cardiovascular and metabolic health. Metrobiotech, a Boston-based company, has generated a pipeline of novel NAD+ precursors, the first of which, MIB-626, is being tested in clinical trials in Boston. Calico has a program to develop NAMPT activators but the program is on hold. Luteolin, a CD38 inhibitor, had positive neuroprotective effects on children with autism but whether NAD+ is responsible for the benefit was not investigated (Tsilioni et al., 2015). SARM1, a promising therapeutic target for axonopathies (Essuman et al., 2017) is in preclinical development at Disarm Therapeutics. PARP1/2 inhibitors improve the health of mice on a HFD (Cerutti et al., 2014). Although, their toxicity may preclude them from use in chronic diseases, there are others with fewer side effects in development (Malyuchenko et al., 2015).
Perspective
Given that NAD+ was discovered over 100 years ago, it is surprising how little we know about its biology, pharmacology, and role in human disease. Though there is much to learn, we know this much: NAD+ boosters seem relatively safe and have a remarkable ability to prevent and treat diseases. Whether these findings will translate to humans is the next big question. Preliminary results in small human clinical trials look promising but there is a long way to go. For NAD+ boosters to be used widely as drugs, it will be important to know their safety profile and understand fundamental aspects of NAD+ biology and physiology, such as the tissue distribution of NAD+ precursors and other NAD boosters, their transport into the cell and into organelles, and the metabolism of NAD+ intermediates in the gut, the bloodstream, on the plasma membrane, and within organelles. There are also practical issues to overcome, such as how to stabilize NAD+ boosters, how to best deliver them and at what dose, what are the best biomarkers and analytical methods, and whether it is best to modulate the degradation or synthesis of NAD+ to achieve the desired efficacy in specific diseases. It is exciting to imagine an NAD+ booster being tested in humans for the ability to increase vitality, reduce all causes of mortality, and extend healthy lifespan. If that happens, more than the discoverers of NAD+ could ever have imagined, NAD+ would truly be the molecule of life.
Content retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6342515/.