NAD+ metabolism and its roles in cellular processes during ageing
Nicotinamide adenine dinucleotide (NAD+) is a coenzyme for redox reactions, making it central to energy metabolism. NAD+ is also an essential cofactor for non-redox NAD+-dependent enzymes, including sirtuins, CD38 and poly(ADP-ribose) polymerases. NAD+ can directly and indirectly influence many key cellular functions, including metabolic pathways, DNA repair, chromatin remodelling, cellular senescence and immune cell function. These cellular processes and functions are critical for maintaining tissue and metabolic homeostasis and for healthy ageing. Remarkably, ageing is accompanied by a gradual decline in tissue and cellular NAD+ levels in multiple model organisms, including rodents and humans. This decline in NAD+ levels is linked causally to numerous ageing-associated diseases, including cognitive decline, cancer, metabolic disease, sarcopenia and frailty. Many of these ageing-associated diseases can be slowed down and even reversed by restoring NAD+ levels. Therefore, targeting NAD+ metabolism has emerged as a potential therapeutic approach to ameliorate ageing-related disease, and extend the human healthspan and lifespan. However, much remains to be learnt about how NAD+ influences human health and ageing biology. This includes a deeper understanding of the molecular mechanisms that regulate NAD+ levels, how to effectively restore NAD+ levels during ageing, whether doing so is safe and whether NAD+ repletion will have beneficial effects in ageing humans.
Nicotinamide adenine dinucleotide (NAD+) is an important coenzyme for redox reactions, making it central to energy metabolism. NAD+ is also an essential cofactor for non-redox NAD+-dependent enzymes, including sirtuins and poly(ADP-ribose) polymerases (PARPs).
NAD+ was first identified for its role in regulating metabolic rates in yeast extracts and later as the major hydride acceptor in redox reactions. This ability of NAD+ to accept a hydride ion, forming its reduced version, NADH, is critical for metabolic reactions of all living life forms and regulates the activity of dehydrogenases involved in multiple catabolic pathways, including glycolysis, glutaminolysis and fatty acid oxidation. The accepted electrons in these reactions are then donated to the electron transport chain to form ATP in eukaryotes. NAD+ can also be phosphorylated to form NADP+, which also acts as a hydride acceptor to form NADPH and is used in protection against oxidative stress and in anabolic pathways that require reducing power, such as fatty acid synthesis.
In addition to energy metabolism, NAD+ is used as a cofactor or substrate by hundreds of enzymes1 and therefore has multiple roles in the regulation of cellular processes and cellular functions, many of which are still being investigated. Links between NAD+ levels and health were established almost a century ago. In 1937, Conrad Elvehjem discovered that the disease pellagra (characterized by dermatitis, diarrhoea and dementia) is caused by a dietary deficiency of niacin and resulted in low NAD+ and NADP+ levels. Recently, low NAD+ levels were linked to multiple disease states, including metabolic and neurodegenerative diseases, and lower NAD+ levels are now known to correlate with ageing in rodents and humans. As a result, there is renewed interest in understanding how NAD+ metabolism impacts the origin of diseases, particularly ageing-related diseases. In this regard, restoring NAD+ levels with the NAD+ precursors nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN) has emerged as an important therapeutic approach to treat age-related diseases and appears to have beneficial effects in vivo, at least in rodent models.
In this Review, we focus on the past 5 years of work in the NAD+ field. In particular, we examine the molecular mechanisms by which NAD+ precursors and ultimately NAD+ levels influence physiology and healthspan in ageing and disease states. Those mechanisms are likely to be complicated and multifactorial, and so we discuss them in multiple sections. First, we review new findings on how the primary NAD+ biosynthetic and degradative pathways are modulated during ageing. Second, we discuss the possible consequences of lower NAD+ levels on molecular processes that are important to ageing-related disease, including DNA repair, regulation of epigenetics and gene expression, regulation of cell metabolism and redox balance. Third, we describe the NAD+-dependent mechanisms in ageing, including metabolic disorders, deregulation of the immune system, cellular senescence and neurodegeneration. Finally, we review the numerous recent high-quality preclinical studies that investigated methods to restore NAD+ levels to treat ageing-related diseases. These include numerous studies using NAD+ precursors, and small-molecule drugs that promote NAD+ biosynthesis. We finish by discussing these different strategies and the results of these studies to outline the prospects of therapies based on modulation of NAD+ levels in promoting human healthspan and lifespan.
Cellular NAD+ metabolism
NAD+ is highly compartmentalized in the cytoplasm, mitochondria and nucleus, which represent its main subcellular pools (Supplementary Box 1). These pools are regulated independently of each other, and consistent with this, the enzymes involved in the biosynthesis or degradation of NAD+ are highly compartmentalized as well2–4. NAD+ is a critical metabolite and coenzyme for multiple metabolic pathways and cellular processes. First, NAD+ reduction is required to maintain energy balance and the redox state of a cell. NAD+ is also continually turned over by three classes of NAD+-consuming enzymes: the NAD+ glycohydrolases, also referred to as NADases (CD38, CD157 and SARM1), the protein deacylase family of sirtuins and PARPs, which have various important cellular functions. These utilize NAD+ as a substrate or cofactor and generate nicotinamide (NAM) as a by-product (FIG. 1). Therefore, NAD+ mediates multiple major biological processes and is always in high demand (Supplementary Boxes 2 and 3). To sustain NAD+ levels, NAM can be recycled back to NAD+ via the NAM salvage pathway (for details see BOX 1). Additionally, some cells, mostly in the liver, can synthesize NAD+ de novo from multiple dietary sources. Thus, NAD+ is constantly synthesized, catabolized and recycled in the cell to maintain stable intracellular NAD+ levels (FIG. 1). However, during ageing, this balance between catabolic and anabolic processes can shift, and NAD+ degradation can outpace the ability of cells to make NAD+ de novo or their ability to effectively recycle or salvage NAM. Furthermore, excess NAM may be catabolized via alternative metabolic pathways (for details see BOX 2), effectively diverting it away from the NAM salvage pathway and further impacting NAD+ levels. Besides their common role as NAD+-consuming enzymes, NAD+ glycohydrolases, sirtuins and PARPs have distinct roles in ageing and age-related diseases. While enhancing the activation of sirtuins has emerged as a way to increase lifespan and healthspan, aberrant activation of PARPs and NAD+ glycohydrolases, such as CD38, may exert the opposite effect and exacerbate ageing phenotypes (see later).
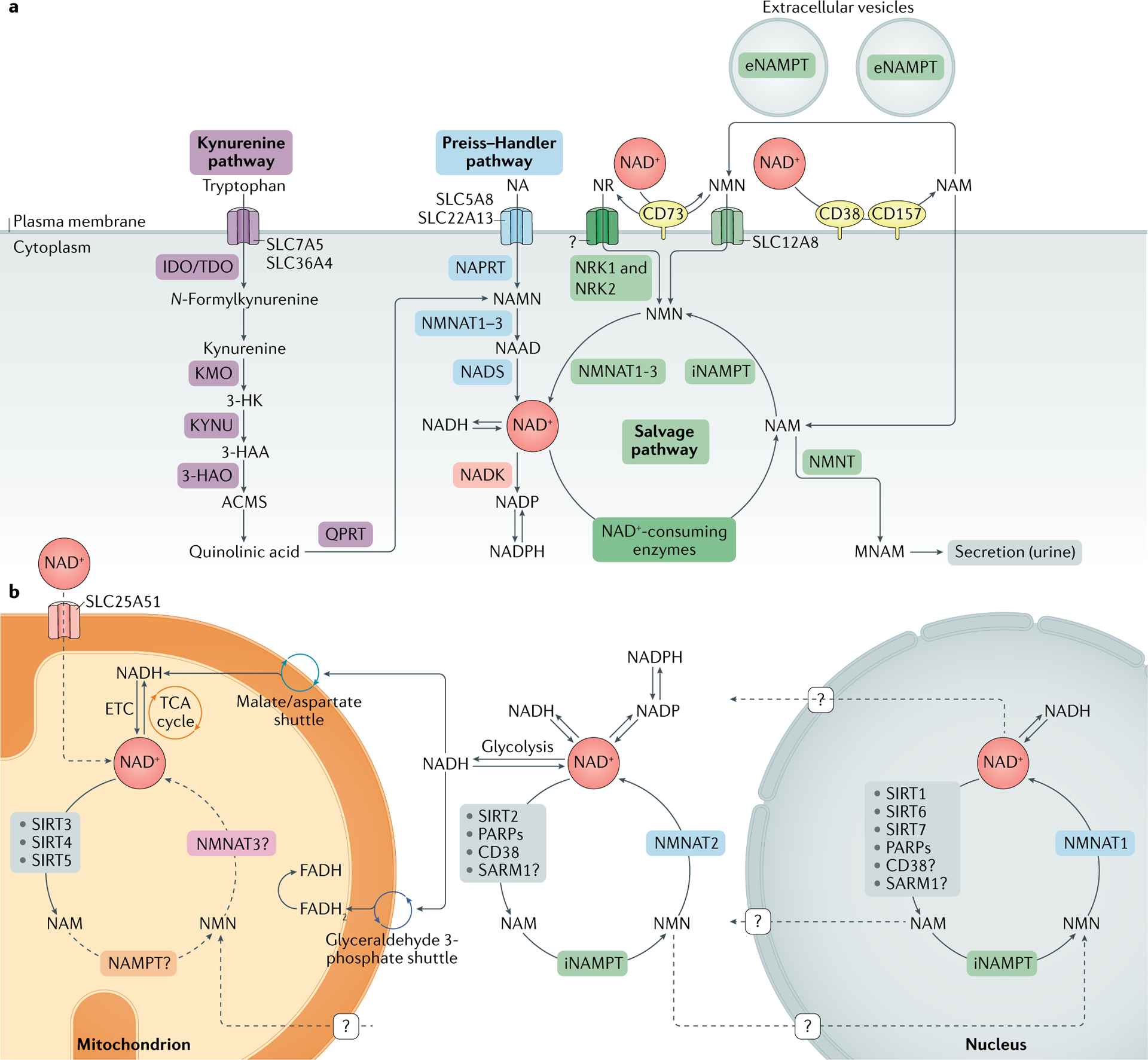
NAD+ metabolism.
a | Nicotinamide adenine dinucleotide (NAD+) biosynthetic pathways. NAD+ levels are maintained by three independent biosynthetic pathways. The kynurenine pathway (or de novo synthesis pathway) uses the dietary amino acid tryptophan to generate NAD+. Tryptophan enters the cell via the transporters SLC7A5 and SLC36A4. Within the cell, tryptophan is converted to N-formylkynurenine by the rate-limiting enzyme indoleamine 2,3-dioxygenase (IDO) or the rate-limiting enzyme tryptophan 2,3-dioxygenase (TDO). N-Formylkynurenine is transformed into l-kynurenine, which is further converted to 3-hydroxykynurenine (3-HK) by kynurenine 3-monooxygenase (KMO) and to 3-hydroxyanthranilic acid (3-HAA) by tryptophan 2,3-dioxygenase (KYNU). The next step is performed by 3-hydroxyanthranilic acid oxygenase (3HAO) to generate α-amino-β-carboxymuconate ε-semialdehyde (ACMS). This compound can spontaneously condense and rearrange into quinolinic acid, which is transformed by quinolinate phosphoribosyltransferase (QPRT) into nicotinamide mononucleotide (NAMN), at which point it converges with the Preiss–Handler pathway. The Preiss–Handler pathway uses dietary nicotinic acid (NA), which enters the cell via SLC5A8 or SLC22A13 transporters, and the enzyme nicotinic acid phosphoribosyltransferase (NAPRT) to generate NAMN, which is then transformed into nicotinic acid adenine dinucleotide (NAAD) by nicotinamide mononucleotide adenylyltransferases (NMNAT1, NMNAT2 and NMNAT3). The process is completed by the transformation of NAAD into NAD+ by NAD+ synthetase (NADS). The NAD+ salvage pathway recycles the nicotinamide (NAM) generated as a by-product of the enzymatic activities of NAD+-consuming enzymes (sirtuins, poly(ADP-ribose) polymerases (PARPs) and the NAD+ glycohydrolase and cyclic ADP-ribose synthases CD38, CD157 and SARM1). Initially, the intracellular nicotinamide phosphoribosyltransferase (iNAMPT) recycles NAM into nicotinamide mononucleotide (NMN), which is then converted into NAD+ via the different NMNATs. NAM can be alternatively methylated by the enzyme nicotinamide N-methyltransferase (NNMT) and secreted via the urine. In the extracellular space, NAM is generated as a by-product of the ectoenzymes CD38 and CD157 and can be converted to NMN by extracellular NAMPT (eNAMPT). NMN is then dephosphorylated by CD73 to nicotinamide riboside (NR), which is transported into the cell via an unknown nucleoside transporter (question mark). NMN can be imported into the cell via an NMN-specific transporter (SLC12A8 in the small intestine). Intracellularly, NR forms NMN via nicotinamide riboside kinases 1 and 2 (NRK1 and NRK2). NMN is then converted to NAD+ by NMNAT1, NMNAT2 and NMNAT3. b | NAD+ metabolism in different subcellular compartments. The NAD+ homeostasis is a balance of synthesis, consumption and regeneration in different subcellular compartments, which are regulated by subcellular-specific NAD+-consuming enzymes, subcellular transporters and redox reactions. NAD+ precursors enter the cell via the three biosynthetic pathways (part a). In the cytoplasm, NAM is converted to NMN by intracellular NAMPT (iNAMPT). NMN is then converted to NAD+ by NMNAT2, which is the cytosol-specific isoform of this enzyme. NAD+ is utilized during glycolysis, generating NADH, which is transferred to the mitochondrial matrix via the malate/aspartate shuttle and the glyceraldehyde 3-phosphate shuttle. The mitochondrial NADH imported via the malate/aspartate shuttle is oxidized by complex I in the electron transport chain (ETC), whereas the resulting reduced flavin adenine dinucleotide (FADH2) from the glyceraldehyde 3-phosphate shuttle is oxidized by complex II. Recently the mammalian NAD+ mitochondrial transporter SLC25A51 was identified, and it has been demonstrated to be responsible for intact NAD+ uptake in the organelle. The salvage pathway for NAD+ in mitochondria has not been fully resolved, but the role of a specific NMNAT isoform (NMNAT3) has been proposed. In the mitochondria, NAD+ is consumed by NAD+-dependent mitochondrial SIRT3–SIRT5, generating NAM. It is still unknown whether NAM can be converted back to NMN or can be converted to NAD+ within the mitochondrion, or whether other precursors can be transported through the mitochondrial membrane to fuel NAD+ synthesis. The nuclear NAD+ pool probably equilibrates with the cytosolic one by diffusion through the nuclear pore; however, the full dynamics are still largely unexplored. A nuclear-specific NMNAT isoform (NMNAT1) has been described and is part of the nuclear NAM salvage NAD+ pathway. MNAM, N1-methylnicotinamide; TCA, tricarboxylic acid.
Box 1 |. Generation of NAD+ via the NAM salvage pathway
The nicotinamide (NAM) salvage pathway generates nicotinamide adenine dinucleotide (NAD+) from the precursor NAM or the upstream NAD+ vitamin precursor nicotinamide riboside (NR) or nicotinamide mononucleotide (NMN) (see FIG. 1a), which can be found in a variety of daily foods such as milk, fruits, vegetables and meat178,199. As NAD+ is not cell permeable, it was thought that all the dietary precursors, including nicotinic acid, NAM, NR and tryptophan, are imported directly into the cells and made available for NAD+ biosynthesis, with the exception of NMN. In support of this, extracellular NMN must be converted to NR by the 5′-nucleotidase CD73 before being imported, thus creating a rate-limiting conversion back to NMN intracellularly by nicotinamide riboside kinase 1 (NRK1)245. A recent study revealed the presence of a specific NMN transporter, SLC12A8, that is highly expressed in the small intestine, suggesting that NMN represents a direct entry point into the NAD+ biosynthetic pathway246. Further studies are necessary to determine the physiological relevance of this transporter to disease, the kinetics and mechanisms of NMN uptake and those of other NAD+ precursors, and their selective expression and effects in each tissue and/or cell in mammals.
NAD+ recycling via the NAM salvage pathway is a fundamental step to restore NAD+ levels after irreversible degradation mediated by the different classes of NAD+-consuming enzymes, including glycohydrolases (CD38, CD157 and SARM1), protein deacylases (sirtuins) and poly(ADP-ribose) polymerases (PARPs). Although the activities and uses of NAD+ differ for each of these enzymes (see FIG. 2), all NAD+-consuming enzymes produce NAM as a by-product of NAD+ degradation; this is converted by the NAM salvage pathway enzyme nicotinamide phosphoribosyltransferase (NAMPT) to NMN. NMN can also be generated from NR by NRK1 and NRK2 (REFS5,245) and converted to NAD+ during the last step of the salvage pathway by the nicotinamide mononucleotide adenylyltransferases NMNAT1, NMNAT2 and NMNAT3 (REFS247,248). Three isoforms of NMNAT have different subcellular localization (NMNAT1, nucleus; NMNAT2, cytosolic face of the Golgi apparatus; NMNAT3, mitochondria) and were reported to regulate NAD+ levels in their respective cellular compartment, but also to influence other intracellular NAD+ stores4,249,250.
NAMPT is ubiquitously expressed and mostly upregulated in processes that depend on high NAD+ levels, such as immune cell activation251 and genotoxic stress252. NAMPT-mediated NAD+ biosynthesis influences cellular metabolism and responses to inflammatory, oxidative and genotoxic stress253,254 by modulating activities of sirtuins and PARP100,253,255. Moreover, NAMPT expression is regulated by the circadian clock machinery (CLOCK–BMAL1) in a feedback loop involving SIRT1, and this correlates with the circadian oscillation of NAD+ levels in vivo10,11. NAMPT exists in two forms in mammals: intracellular NAMPT (iNAMPT) and extracellular NAMPT (eNAMPT). To function as an NAD+ biosynthetic enzyme, NAMPT forms homodimers256. Many cell types produce eNAMPT130, and its secretion is actively regulated by SIRT1 or SIRT6-mediated deacetylation257 in adipose tissue and cancer cell lines258, respectively. More recently, it was shown that eNAMPT is carried in extracellular vesicles in the blood circulation of mice and humans (see FIG. 1a) and it is responsible for enhancing the intracellular NAD+ biosynthesis in primary hypothalamic neurons259. In addition to its role in regulating NAM salvage, eNAMPT was previously identified as a presumptive cytokine (also known as pre-B cell colony-enhancing factor (PBEF))260. The levels of eNAMPT monomer in the serum are increased in a number of immunological and metabolic disorders261,262. However, it is unclear whether the enzymatic activity of NAMPT is necessary for its cytokine-like function. Therefore, discriminating between monomer and dimer forms of eNAMPT could be a way to resolve the current controversy about enzymatic activity263 and function of eNAMPT. Collectively, these studies suggest that eNAMPT acts to maintain NAD+ in distant cells in an autocrine fashion and, remarkably, also acts as an endocrine signal to influence NAD+ levels and inflammation in distant tissues.
Box 2 |. Metabolic catabolism of NAD+ via NNMT and NADK
Nicotinamide N-methyltransferase (NNMT) is an enzyme that uses S-methyladenosine (SAM) as a methyl donor to methylate the ring nitrogen of nicotinamide (NAM), converting it to N1-methylnicotinamide (MNAM) (see FIG. 1a). MNAM can be further oxidized via aldehyde oxidase to produce N1-methyl-2-pyridone-5-carboxamide and N1-methyl-4-pyridone-3-carboxamide, which along with MNAM are secreted in the urine. NNMT conversion of NAM to MNAM effectively diverts NAM from being recycled back to nicotinamide adenine dinucleotide (NAD+) by the NAM salvage pathway and affects global NAD+ levels. Thus, there has been growing interest in the role of NNMT as a key regulator of NAD+ levels during disease states, such as obesity, cancer and ageing264. For example, NNMT expression increases in visceral white adipose tissue and liver during obesity, and appears to have mostly negative consequences265–268. This includes the depletion of the methyl donor SAM and NAD+ in white adipose tissue and the liver in response to a high-fat diet. As a result, increased NNMT activity leads to reduced methylation of the promoters of genes involved in fibrosis and aberrant gene expression of metabolic and inflammatory genes266. Thus, NNMT appears to be a critical metabolic enzyme linking NAD+ metabolism to control of gene expression via its ability to regulate levels of bioactive molecules, such as consumption of NAD+ and SAM, to produce MNAM.
The role of NNMT in regulating NAD+ suggests that NNMT is important in ageing. In support of this, the Caenorhabditis elegans homologue of NNMT, ANMT-1, regulates the production of MNAM. In worms, MNAM serves as a substrate for the aldehyde oxidase GAD-3, which produces hydrogen peroxide, which, in turn, acts as a hormesis signal to promote longevity. Worms lacking anmt-1 no longer benefit from sir-2.1-dependent lifespan extension191. These data suggest that consumption of NAD+ via NNMT is beneficial for lifespan extension at least in C. elegans. However, it is unclear what role NNMT activation plays in mammalian ageing, although NNMT expression and activity do appear to increase in rodents as they age. For example, our group recently saw NNMT gene expression increase in the hepatocytes of ageing mice45, and a similar increase was observed in hepatocytes of older mice fed a high-protein diet269. Additionally, treating old mice with an NNMT inhibitor activated senescent muscle stem cells and promoted muscle regeneration during ageing270. Thus, in contrast to C. elegans, in mammals early evidence suggests that NNMT may promote ageing-related diseases. Thus, targeting NNMT may provide a viable therapeutic avenue to treat ageing-related diseases.
Another metabolic fate of NAD+ is direct phosphorylation by NAD+ kinase (NADK) to produce NADP(H), which has a key role as a major source of reducing power that regulates intracellular redox balance and anabolic processes, such as lipogenesis. A recent study using in vitro isotopic tracing label incorporation into NADP(H) showed that NADK accounts for 10% of NAD+ consumption5. However, the total NADP+ pool is 20 times less than the NAD+ pool. Furthermore, in conditions where NAD+ levels decline, such as in ageing, NADP+ levels also decline. Thus, NADP+ levels appear to be directly linked to NAD+ levels and rise and fall in tandem, making it unlikely that NADK is a major NAD+ sink. However, NADK has recently been shown to be a direct target of AKT kinase, phosphorylating the residues Ser44, Ser46 and Ser48 and leading to enhanced activation of NADK271. Thus, given that AKT signalling is abnormally increased during ageing272, aberrant activation of NADK may account for some of the decline in NAD+ levels during ageing. Therefore, further studies will be needed to determine the role of NADK in regulating NAD+ and NADP+ pools in ageing and ageing-related diseases.
NAD+ biosynthetic pathways
NAD+ can be made de novo from l-tryptophan via the kynurenine pathway or from vitamin precursors, such as nicotinic acid (NA), via the Preiss–Handler pathway (FIG. 1a). In addition to NAD+, the kynurenine pathway also utilizes l-tryptophan to produce kynurenic acid, serotonin and picolinic acid, among other bioactive molecules. Notably, the relative contribution of the de novo synthesis pathway to NAD+ levels is still not well understood. Outside the liver, most cells do not express the full array of enzymes necessary to convert tryptophan to NAD+ by the kynurenine pathway. Most tryptophan is metabolized to NAM in the liver, where it is released into the serum, taken up by peripheral cells and converted to NAD+ by the NAM salvage pathway5. Additionally, under some circumstances, immune cells, such as macrophages, also make NAD+ from tryptophan (discussed later)6. Thus, besides the liver, the de novo biosynthetic pathway seems to be a more indirect mechanism contributing to system-wide NAD+ levels, with most NAD+ coming from the NAM salvage pathway (BOX 1).
NAD+ consumption routes
Consumption by sirtuins.
Since their discovery, the sirtuins have received much attention because they regulate key metabolic processes, stress responses and ageing biology7. The mammalian sirtuin family is composed of seven genes and proteins (SIRT1–SIRT7) with different subcellular localizations (nucleus for SIRT1 and SIRT6; nucleolus for SIRT7; mitochondria for SIRT3, SIRT4 and SIRT5; and cytosol for SIRT1, SIRT2 and SIRT5) (FIG. 1b), enzymatic activities and downstream targets. The sub-cellular localization of these NAD+-dependent enzymes emphasizes how local fluctuations of intracellular NAD+ pools, which are themselves regulated by sirtuins, may selectively impact organelle-specific sirtuin functions and cellular metabolism.
Sirtuins are continuously active in our cells. SIRT1 and SIRT2 seem to be responsible for about one third of the total NAD+ consumption under basal conditions5. Moreover, a rise of NAD+ levels is strongly correlated with sirtuin activation during fasting and caloric restriction8,9. It is also worth noting that sirtuin activity is coupled to the circadian clock, whereby SIRT1 and SIRT6 regulate the activity of the core clock transcription factors and downstream circadian transcriptome8. In addition, SIRT1 and a key enzyme of the NAD+ salvage pathway, nicotinamide phosphoribosyltransferase (NAMPT) (BOX 1), are key players in the circadian regulation of NAD+ levels, and NAMPT is regulated by the circadian clock, which provides a feedback loop, resulting in circadian oscillation of NAD+ levels10,11.
Early studies in yeast demonstrated roles of sirtuins in gene silencing, which in 2000 was attributed to their activity as NAD+-dependent deacetylases12. Sirtuin enzymatic activity was initially reported to encompass mainly removal of an acetyl group from lysine residues of target proteins in a two-step process: first NAD+ is cleaved to NAM and ADP-ribose, and second, the acetyl group on the target protein is transferred to ADP-ribose, allowing the formation of the intermediate peptidyl-ADP-ribose. Acetyl-ADP-ribose is subsequently released (FIG. 2a). Additionally, some members of the sirtuin family mediate non-acetyl lysine acylations (for example, succinylation, malonylation and fatty acid acylation)13–15, the cellular functions of which are so far poorly understood16. SIRT4 and SIRT6 also function as ADP-ribosyltransferases, indicating that sirtuins contribute to cell regulation also through mechanisms beyond modulation of protein acetylation, which require further investigation17,18.
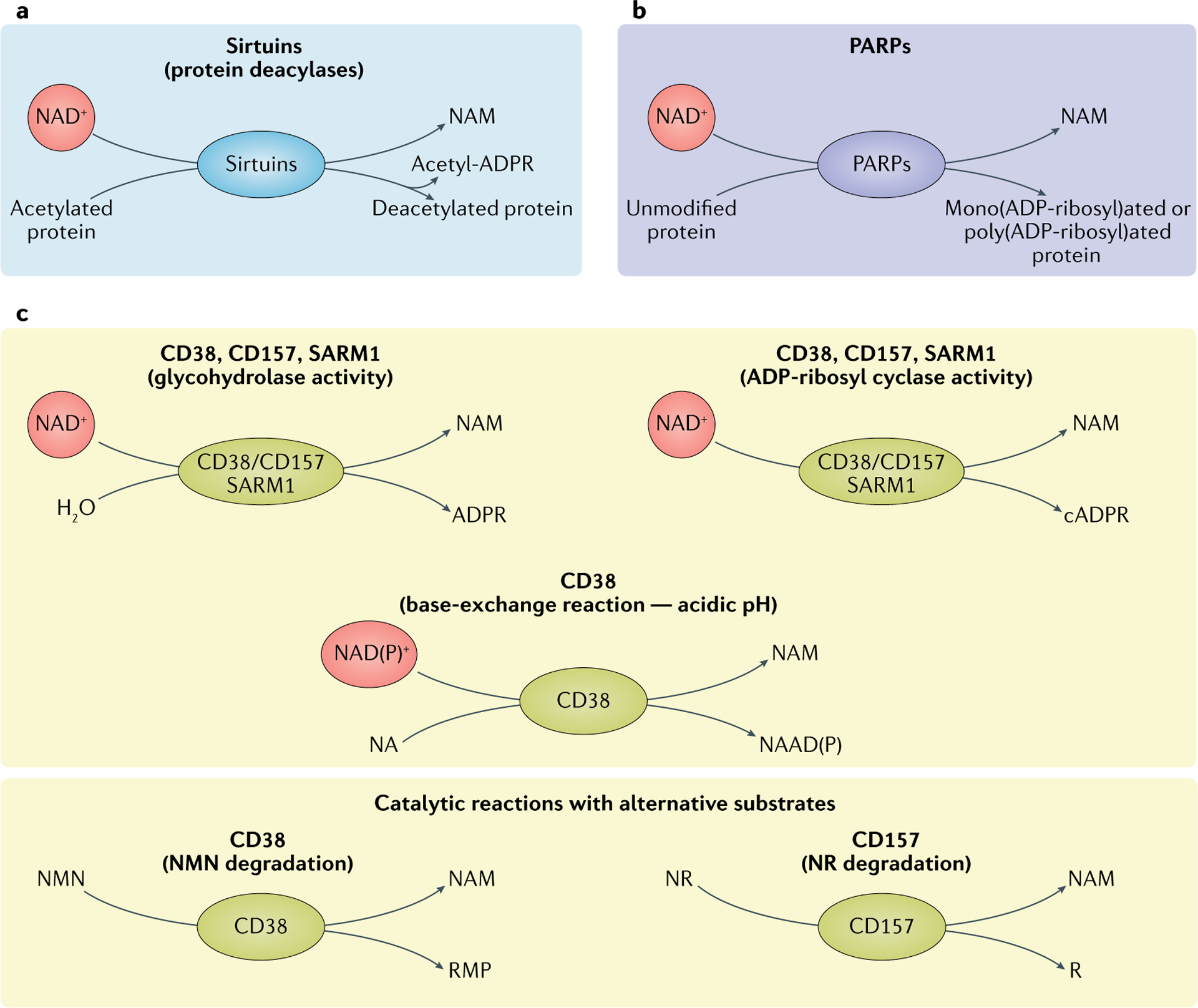
Three main classes of NAD+-consuming enzymes.
a | Sirtuins remove acyl groups from lysine residues on target proteins using nicotinamide adenine dinucleotide (NAD+) as a co-substrate. NAD+ is cleaved, generating nicotinamide (NAM) and ADP-ribose, where ADP-ribose serves as an acyl group acceptor, generating acetyl-ADP-ribose (acetyl-ADPR). b | Poly(ADP-ribose) polymerases (PARP1–PARP3) use NAD+ as a co-substrate to mono(ADP-ribosyl)ate or poly(ADP-ribosyl)ate target proteins, generating NAM as a by-product. c | Reactions of NAD+ glycohydrolases and cyclic ADP-ribose (cADPR) synthases (CD38, CD157 and SARM1). The main catalytic activity of this group of proteins is the hydrolysis of NAD+ to NAM and ADP-ribose. To a lesser extent, CD38, CD157 and SARM1 have ADP-ribosyl cyclase activity, generating NAM and cADPR from NAD+. In acidic conditions, CD38 can also perform a base-exchange reaction, swapping the NAM of NAD(P)+ for nicotinic acid (NA), generating nicotinic acid adenine dinucleotide (phosphate) (NAAD(P)). CD38 and CD157 are reported to be able to use alternative substrates in their catalytic reactions. CD38 can degrade NMN to NAM and ribose monophosphate (RMP), while CD157 can degrade NR, generating NAM and ribose (R). NR, nicotinamide riboside.
Although measuring NAD+ levels and sirtuin activity in subcellular compartments has been hampered by technological challenges, owing to recent technical advancements (Supplementary Box 4) we now have some insights into distinct cellular roles of these enzymes. Specifically, recent evidence has shown that nuclear SIRT1, SIRT6 and SIRT7 are critical regulators of DNA repair and genome stability and that mitochondrial SIRT3, SIRT4 and SIRT5 and nuclear SIRT1 regulate mitochondrial homeostasis and metabolism7. In apparent contrast with its role in promoting mitochondrial biogenesis through peroxisome proliferator-activated receptor-γ co-activator 1α deacetylation19–21, SIRT1 has also been implicated in turnover of defective mitochondria by mitophagy22,23. However, in both cases SIRT1 seems to be a key factor in mitochondrial quality maintenance9. Overall, sirtuins have emerged as key actors in the efforts to understand and characterize how NAD+ levels affect cellular homeostasis in a wide variety of cellular processes that impact ageing (Supplementary Box 2), and boosting their activity is a key focus of therapies aimed at counteracting ageing.
Consumption by PARPs.
The human PARP protein family is composed of 17 proteins characterized by poly(ADP-ribosyl) polymerase or mono(ADP-ribosyl) polymerase activity24. Briefly, the PARP-mediated cleavage of NAD+ produces NAM and ADP-ribose as by-products, in which ADP-ribose is added as single or covalently linked polymers to PARP itself and other acceptor proteins, in a process called ‘poly(ADP-ribosyl)ation’ (PARylation) (FIG. 2b). Among all the PARPs, only PARP1, PARP2 and PARP3 localize to the nucleus (FIG. 1b) in response to early DNA damage and have a key role in DNA damage repair25–27 (Supplementary Box 2).
PARP1 is the best-characterized member of the group, and it alone is responsible for about 90% of total PARP activity at least in response to DNA damage28,29. On activation, PARP1 PARylates itself along with histones and other proteins that act as a scaffold to recruit and activate other DNA repair enzymes and proteins to the lesion site to initiate DNA repair30. Owing to high PARP1 activity, DNA damage is associated with massive NAD+ consumption. PARP1, in its role as an NAD+ responsive signalling molecule, has been widely associated with the ageing process. However, PARP1 is one of the major NAD+ consumers not only in cells with acute DNA damage but also under normal and other pathophysiological conditions5, supporting a key role of PARP1 in regulating NAD+ homeostasis. For example, in mice fed a high-fat diet, those that were treated with PARP inhibitors or lacking PARP1 and PARP2 had increased NAD+ levels, increased SIRT1 activity and improved mitochondrial function, and were protected from insulin resistance31,32. The strong correlation among PARP1 activation, decline in NAD+ levels and inhibition of SIRT1 activity is observed in patients with xeroderma pigmentosum group A, as well as progeroid diseases, ataxia telangiectasia and Cockayne syndrome. Notably, treatment of mice with Cockayne syndrome with PARP1 inhibitors or NAD+ supplements promoted lifespan extension and ameliorated the severe phenotypes caused by PARP1 hyperactivation, providing strong evidence that the negative consequences downstream of PARP1 activation are mediated by dysregulation of NAD+ homeostasis in response to extensive DNA damage and genotoxic stress33. Of note, the ability of PARP1 to antagonize SIRT1 activity is most likely because these enzymes are localized to the same cellular compartment — in this case, the nucleus — and compete for the same NAD+ pool. However, since PARP1 has lower Km and higher Vmax with regard to NAD+ than SIRT1, PARP1 likely outcompetes SIRT1 for NAD+ as a result of its greater binding affinity and faster kinetics34.
PARP2 is structurally related to PARP1. They share a similar catalytic domain required for several cellular processes, including DNA repair and transcriptional regulation, and account for about 10% of PARP activity35. PARP2 activity may also influence NAD+ bioavailability25,26. Like Parp1-knockout mice, Parp2-knockout mice show enhanced SIRT1 activity and improved metabolic function, and are protected from high-fat diet-induced obesity36. PARP3 is important in DNA repair as well27, suggesting a large overlap and potential redundancy in PARP1, PARP2 and PARP3. The functions of the other PARPs (PARP4–PARP17) in NAD+ homeostasis and global metabolism in cells or organs have not been fully determined, but they are thought to be less important in modulating intracellular NAD+ levels. Overall, targeting PARPs, and in particular PARP1, is a promising therapeutic strategy in the ageing field. However, more studies are needed to fully understand the contribution of PARPs to the age-related decline in NAD+ levels.
Consumption by CD38 and CD157.
CD38 and CD157 are multifunctional ectoenzymes with both glycohydrolase and ADP-ribosyl cyclase activities. The glycohydrolysis of NAD+ is the primary catalytic reaction that cleaves the glycosidic bond within NAD+ to generate NAM and ADP-ribose, whereas the ADP-ribosyl cyclase activity generates cyclic ADP-ribose (FIG. 2c). CD38 also performs a base-exchange reaction, by swapping the NAM of NAD(P)+ for NA in acidic conditions and generating nicotinic acid adenine dinucleotide (phosphate) (NAAD(P))37 (FIG. 2c). Of note, cyclic ADP-ribose, NAAD(P) and ADP-ribose are all key Ca2+-mobilizing second messengers, illustrating the pivotal role of CD38 in activating Ca2+ signalling and modulating essential cell processes, such as immune cell activation, survival and metabolism38–40. Importantly, other than NAD+ and NADP+, NMN is emerging as an alternative substrate of CD38 (REFS41,42), whereas CD157 consumes NR as an alternative substrate43,44 (FIG. 2c). Thus, targeting CD38 and CD157 with small-molecule inhibitors may make these commonly used NAD+ precursor metabolites more efficient in restoring NAD+ levels in ageing individuals. Very little is known about the purpose of CD157 enzymatic functions in cellular biology or ageing. However, recent evidence shows that, like CD38, CD157 is upregulated in ageing tissues45 and may have a role in ageing-related diseases, such as rheumatoid arthritis and cancer46.
Although CD38 and CD157 are genetically homologous and are members of the same enzymatic family, their structure, localization and role in diseases differ. CD38 is a transmembrane protein with a type ii orientation and/or a type III orientation identified in the late 1970s as a T cell activation marker but now known to be ubiquitously expressed, particularly during inflammation47,48. CD157 is a glycophosphatidylinositol-anchored protein, which was first identified in the myeloid compartment of the haematopoietic system49. However, CD157 is also expressed by other cells, including B cell progenitors, Paneth cells and endothelial cells in the gut, pancreas and kidney50.
Besides their enzymatic function, CD38 and CD157 act as cell receptors. For example, CD38 is an adhesion receptor interacting with CD31 to mediate immune cell trafficking and extravasation through the endothelium51,52. The CD38–CD31 axis seems to promote a proliferative response in chronic lymphocytic leukaemia lymphocytes, suggesting a detrimental role of CD38 in blood cancers53,54 (see Supplementary Box 3). However, very little is known about the functional consequences of CD38–CD31 interaction. Additionally, CD38 is thought to mediate immunity through antimicrobial functions, given that a major phenotype of Cd38-knockout mice is their inability to mount immune responses to bacteria55. It is still unknown whether the CD38 enzymatic function is necessary for its antibacterial functions and to what extent these functions depend on NAD+. However, it is plausible that the role of CD38 in antimicrobial resistance is related to sequestering NAD+ or NAD+-related metabolites from bacteria that require NAD+ for survival and growth56,57.
The role of CD157 as a receptor is still not fully explored. Several lines of evidence suggest that CD157 activation by specific monoclonal antibodies promotes trafficking of neutrophils and monocytes58. Moreover, CD157 interacts with integrins to form a receptor recognized by scrapie-responsive gene 1 protein (SCRG1), promoting self-renewal, migration and osteogenic differentiation of human mesenchymal stem cells43. However, whether these receptor functions are relevant to NAD+ metabolism remain unclear.
Consumption by SARM1.
SARM1 was only very recently assigned to the NAD+ glycohydrolase and cyclase family along with CD38 and CD157. The enzymatic activity of SARM1 (FIG. 2c) relies on the Toll/interleukin receptor (TIR) domain that was previously not known to possess catalytic activity and generally to be involved in protein–protein interaction59. Whether SARM1 regulates NAD+ under physiological conditions and to what extent is still unknown. However, SARM1-mediated NAD+ degradation plays a key role in axonal degeneration after axonal injury60 (see the section Neurodegeneration). SARM1 is primarily expressed in neurons and promotes neuronal morphogenesis and inflammation61,62; however, SARM1 is also expressed by immune cells such as macrophages and T lymphocytes, and regulates their functions63–65. SARM1 was originally discovered as a negative regulator of the innate immune response via direct interaction with TRIF (TIR domain-containing adapter protein inducing interferon-β), downstream of Toll-like receptor signalling63. However, the immunoregulatory role of SARM1 remains largely uncharacterized. Although SARM1 was originally reported to be required for chemokine CCL5 production in macrophages66, recent evidence revealed that SARM1 is not expressed in macrophages and that the observed chemokine phenotype was due to a background effect of the Sarm1-knockout mouse strain67. Despite its controversial role in immune cells, SARM1 plays an undisputed key role in axonal degeneration and is emerging as a therapeutic target to prevent or ameliorate neurodegenerative diseases and traumatic brain injuries (as discussed in the section Neurodegeneration).
Cellular roles of NAD+
Beyond the main groups of NAD+-
Beyond the main groups of NAD+-consuming enzymes discussed so far, NAD+ is widely used as a cofactor or substrate for biochemical reactions, with more than 300 enzymes1 relying on NAD+ for their activity. Accordingly, NAD+ is a mediator of key cellular functions and adaptation to metabolic needs. Some of these critical cellular processes include metabolic pathways, redox homeostasis, maintenance and repair of DNA to safeguard genomic stability, epigenetic regulation and chromatin remodelling, and autophagy. Collectively, these functions are important for maintaining systemic health and homeostasis. However, during ageing, declining NAD+ levels can impinge on these processes and exacerbate ageing-related diseases (FIG. 3; Supplementary Box 2).
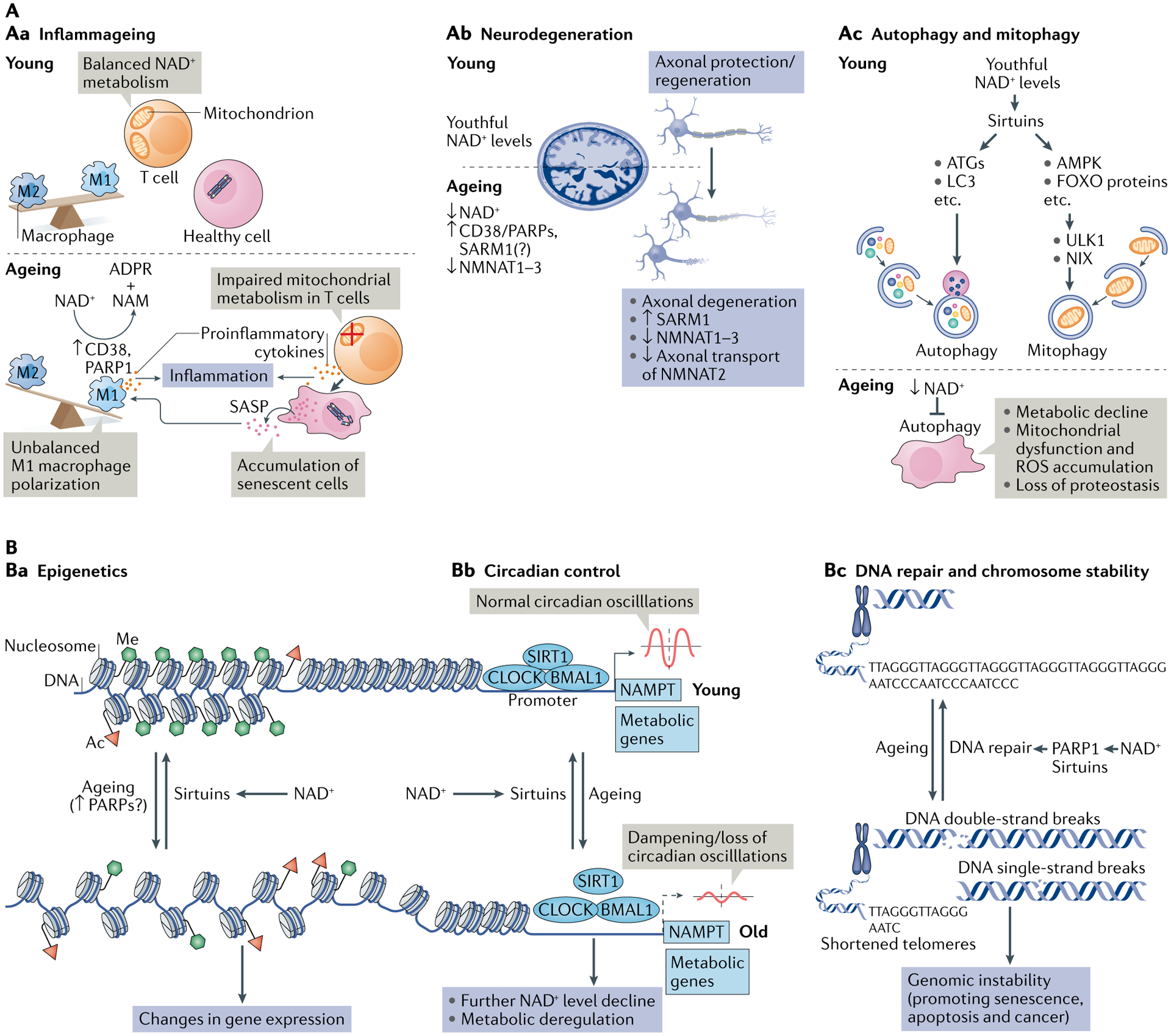
NAD+ metabolism in ageing.
A | Decreased nicotinamide adenine dinucleotide (NAD+) levels have been implicated in various processes associated with ageing (see also Supplementary Box 2). Aa | Ageing is associated with aberrant proinflammatory immune cell activation or ‘inflammageing’, leading to sustained low-grade inflammation. This is caused in part by the accumulation of senescent cells, which via a senescence-associated secretory phenotype (SASP) promote the phenotypic polarization of macrophages towards a proinflammatory M1 state, thereby driving inflammation. There is evidence that in response to the SASP in these macrophages expression of the NAD+-consuming enzymes CD38 and poly(ADP-ribose) polymerases (PARPs) increases, leading to NAD+ level decline, and that these mechanisms importantly contribute to the decrease of NAD+ levels in ageing. In addition, it has been shown that in aged T cells mitochondrial function declines, and this leads to increased secretion of proinflammatory cytokines that promote the state of inflammation and also induce senescence. Ab | Axonal degeneration, which is a precursor to many age-related neuronal disorders, is characterized by rapid NAD+ depletion. During normal physiological conditions, NAD+ biosynthetic enzymes, nicotinamide mononucleotide adenylyltransferases (NMNATs), are protective against axonal degeneration, and their expression supports maintenance of axons and prevents neurodegeneration. In particular, NMNAT2 is an important survival factor in axons, to which it needs to be constantly delivered from the soma — where it is synthesized — to account for its rapid turnover, and these transport processes are disturbed during axonal degeneration. Moreover, the NAD+-consuming enzyme SARM1 is activated by axonal injury and mediates axonal degeneration by promoting NAD+ degradation. Ac | Autophagy is a key cellular catabolic process that allows cells to adapt to variable nutrient availability and serves in cellular quality control, allowing removal of defective organelles and proteins. Autophagy is regulated downstream of NAD+ levels via sirtuins (mostly SIRT1). Decline of NAD+ levels reduces overall autophagic flux as well as selective removal of mitochondria via mitophagy, suggesting that defective autophagy can be a consequence of NAD+ depletion during ageing, contributing to cell dysfunction. B | Because NAD+ is a cofactor for various enzymes, loss of NAD+ impacts a plethora of cellular processes. For example, NAD+ is required for the activity of epigenetic regulators such as SIRT1, and decline in its level causes changes in histone modifications, thereby affecting chromatin organization and function in gene expression. There is evidence that the ageing-associated loss of NAD+ is related to increased expression of PARPs, which can be caused by increased levels of DNA damage and the need for DNA repair during ageing (panel Ba). NAD+ also affects transcriptional activity of the core clock components CLOCK and BMAL, thereby regulating circadian expression of important metabolic genes as well as nicotinamide phosphoribosyltransferase (NAMPT), which in turn is required for circadian oscillation in NAD+ levels (panel Bb). Decreased NAD+ levels also interfere with the activity of PARPs and sirtuins in DNA repair, leading to genomic instability: a hallmark of ageing and cancer (panel Bc). ADPR, ADP-ribose; ATG, autophagy-related protein; FOXO, forkhead box protein O; ROS, reactive oxygen species.
NAD+-dependent mechanisms in ageing
During ageing, NAD+ levels decline, and many enzymes associated with NAD+ degradation and biosynthesis are altered. The relationship between NAD+ and the 10 hallmarks of ageing was extensively reviewed68 (see also Supplementary Table 1). Additionally, this decline in NAD+ levels during ageing has been linked to the development and progression of ageing-related diseases, including atherosclerosis, arthritis, hypertension, cognitive decline, diabetes and cancer3. In this section, we focus on the major cellular processes that influence or are influenced by ageing, such as metabolic dysfunction, DNA repair failure and genomic instability, inflammageing, cellular senescence and neurodegeneration, and discuss their regulation by NAD+ levels. All these processes and the age-related disorders associated with them are likely to benefit from NAD+ repletion. Restoring NAD+ levels with dietary precursors and targeting NAD+ degradation enzymes with small-molecule inhibitors has emerged as a potential therapeutic strategy to restore NAD+ levels, thereby providing opportunities for alleviating age-related decline and diseases (FIG. 4) (see also the next section).
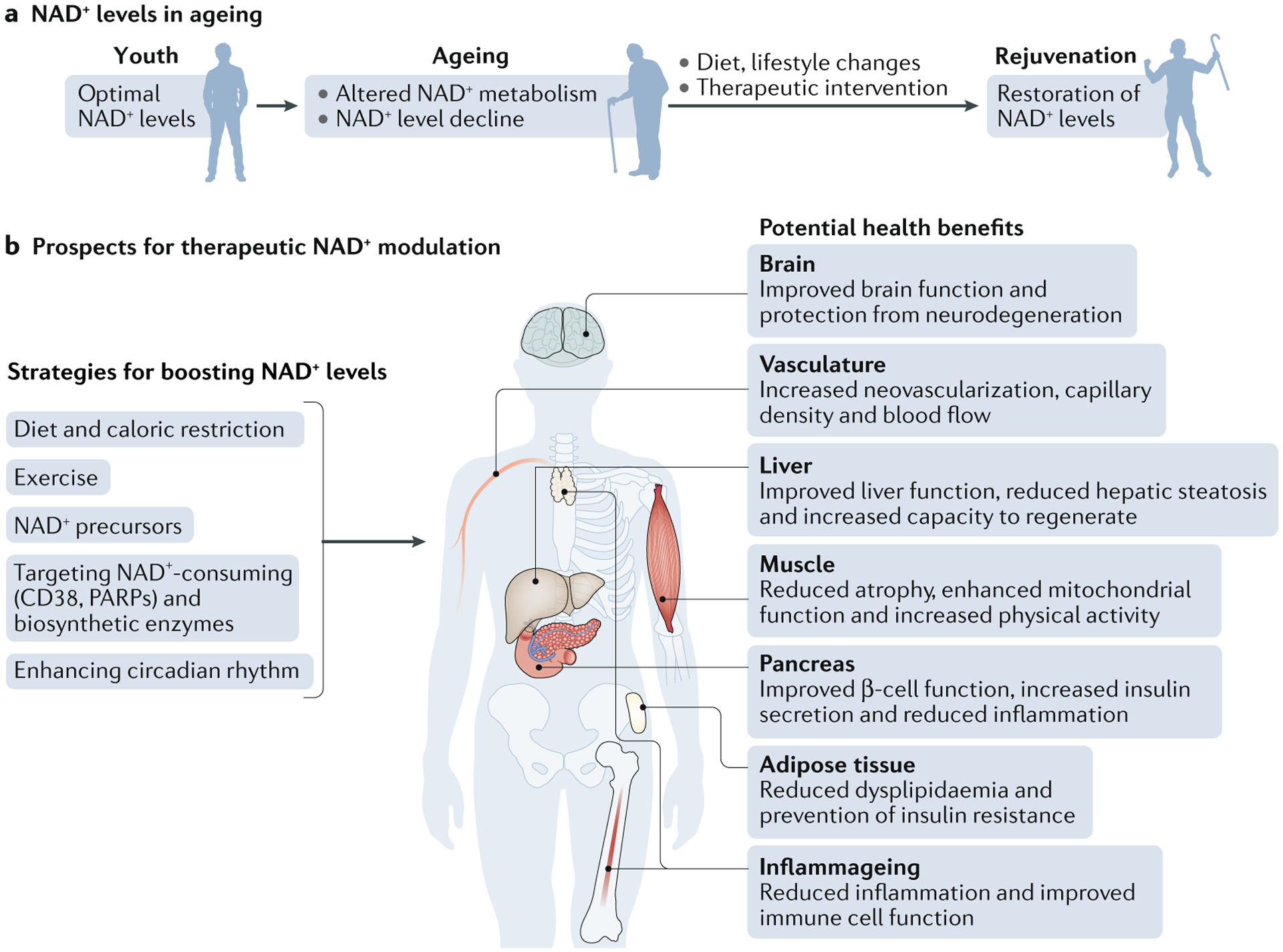
Therapeutic approaches to restore NAD+ levels and their impact on health.
Ageing is associated with decreased nicotinamide adenine dinucleotide (NAD+) levels that promote or exacerbate ageing-related diseases. Thus, restoring NAD+ levels has emerged as a therapeutic approach to prevent and treat ageing-related diseases and to restore health and vigour during the ageing process (part a). Some potential strategies that boost NAD+ levels include lifestyle changes, such as increasing exercise, reducing caloric intake, eating a healthy diet and following a consistent daily circadian rhythm pattern by conforming to healthy sleeping habits and mealtimes. Another approach is the use of small-molecule inhibitors or activators to boost NAD+ biosynthesis and the use of dietary supplements, including NAD+ precursors, such as nicotinamide mononucleotide and nicotinamide riboside. All of these approaches promote tissue NAD+ levels and are beneficial for health. These include improved tissue and organ function, protection from cognitive decline, improved metabolic health, reduced inflammation and increased physiological benefits, such as increased physical activity, which may collectively extend patient healthspan and potentially lifespan (part b).
Metabolic dysfunction
Obesity is a growing epidemic worldwide69. Individuals with obesity are more likely to develop metabolic disease characterized by increased adiposity, insulin resistance, high blood glucose levels, high blood pressure and dyslipidaemia. As a result, these individuals are at higher risk of developing type 2 diabetes, cardiovascular disease, non-alcoholic fatty liver disease, atherosclerosis, stroke and cancer70. Accordingly, obesity accelerates and exacerbates ageing, and has been associated with shortened lifespan71. Over the past two decades growing evidence has accumulated that targeting NAD+ metabolism may provide therapeutic benefits and help treat metabolic disease and ageing3.
NAD+ was first discovered by regulating the metabolic rates of yeast extracts, and thus the links between NAD+ and metabolism have been known for almost a century. We now know that NAD+ lies at the heart of metabolism and regulates metabolic flux of multiple metabolic pathways (for details see BOX 1 and Supplementary Box 2). Accordingly, NAD+ homeostasis is required for the proper function of various metabolic tissues, including fat, muscle, intestines, kidneys and liver (reviewed in72). However, another key discovery was made more recently in Saccharomyces cerevisiae, showing that the longevity protein Sir2 (the yeast sirtuin) exhibited its lifespan-promoting effects in an NAD+-dependent manner12,73,74. This breakthrough finding suggested that Sir2 activity may be linked to metabolic status. In support of this hypothesis, it has now been demonstrated in mammalian systems that lifespan-extending metabolic manipulations, such as exercise75–77, caloric restriction78, time-restricted feeding and a ketogenic diet79, as well as healthy circadian rhythms1,11 (including regular sleep patterns80), work in part by increasing NAD+ levels, which leads to the activation of sirtuins.
Altering or disrupting metabolic status, for example as a consequence of a high-fat diet81,82, postpartum weight loss83 and disruption of the circadian rhythm1,83, can lead to lower NAD+ levels, and thereby reduced activity of sirtuins as well as other NAD+-dependent cellular processes (see Supplementary Box 2 for details). Conversely, increasing cellular NAD+ levels has recently been shown to reduce reductive stress and drive the directionailty of metabolic reactions84. Additionally, higher NAD+ levels can promote the deacetylase and deacylase activity of both nuclear SIRT1 and mitochondrial SIRT3, which regulate mitochondrial function and protect from high-fat diet-induced metabolic disease42,81,85–88.
Taken together, these key studies have provided strong evidence and the rationale that targeting NAD+ degradation pathways or boosting NAD+ levels can influence metabolic processes and can be protective against metabolic disease. In further support of this model, numerous studies have now shown that mouse models including Parp1-knockout and Cd38-knockout mice, or mice treated with PARP or CD38 inhibitors, have supraphysiological levels of NAD+. They are also protected from obesity, have increased metabolic rates and have (relatively) normal glucose metabolism during high-fat diet conditions and during ageing42,89–91. Additionally, mice receiving a high-fat diet have increased inflammation, which drives decreased expression of NAMPT, and reduced activity of the NAD+ salvage pathway82 (BOX 1), providing a potential mechanistic explanation for why NAD+ levels decline during obesity. In line with this mechanism, mice with adipocyte-specific deletion of NAMPT have lower levels of NAD+ in their fat tissues, increased insulin resistance and increased metabolic dysfunction, which can be rescued with NMN supplementation92. Additionally, multiple recent studies have shown that NAD+ supplements (NR and NMN), which can restore low NAD+ levels associated with ageing, are also protective against obesity in rodent models81,82,92, suggesting that these supplements could be explored as therapeutics to restore metabolic health in human patients with obesity. Several recent clinical trials have begun to investigate the efficacy of NAD+ precursors in human patients with obesity to improve metabolic health and glucose metabolism. So far, most of these studies have been done in healthy individuals (see TABLE 1) to test the effective doses of NR and NMN that can safely boost NAD+ levels. However, two recent randomized and double-blind studies treated patients with over-weight and obesity with NR for 6 weeks and 12 weeks93,94. Unfortunately, while NR could effectively increase NAD+ levels in these individuals94, participants in neither study showed any signs of weight loss, increased insulin sensitivity or enhanced mitochondrial function, among other metabolic parameters studied. Thus, it is still unclear whether targeting NAD+ metabolism can be effective in treating metabolic disease in individuals with obesity or ageing individuals.
Table 1 |
Preclinical studies in mouse models of human diseases using NAD+-boosting strategies
| Mechanism of action | Compound | Model | Outcome | Refs |
|---|---|---|---|---|
| NAD+ precursors | Nicotinamide | High-fat diet-fed and standard diet-fed male C57BL/6J wild-type mice | Prevents diet-induced hepatosteatosis and improves glucose metabolism and redox status in liver. Increases pentose phosphate pathway and reduces protein carbonylation. No change in lifespan | 200 |
| Alzheimer disease-relevant 3xTgAD mice | Increases of antioxidant levels, autophagy-lysosome clearance and oxidative stress resistance/mitochondrial function/integrity. Decreases of Aβ peptide and phosphorylated tau levels. Improvements in neuronal plasticity/cognitive function | 233 | ||
| NMN | Male C57BL/6N wild-type mice | Inhibits age-induced weight gain, increases insulin sensitivity, plasma lipid levels, physical activity and energy expenditure and improves muscle mitochondrial function | 199 | |
| C57BL/6J wild-type mice | Enhances skeletal muscle mitochondrial oxidative metabolism in aged mice | 201 | ||
| Male Long-Evans rats Ischaemia-reperfusion or cisplatin-induced acute kidney injury in Sirt1+/− mice, C57BL/6 mice and 129S2/Sv mice |
Improves mitochondrial function, decreases inflammation, improves physiologic reserve and decreases mortality, despite having no major effect on blood pressure or oxidative damage Protects renal function from cisplatin-induced injury in wild-type mice but not in Sirt1+/− mice |
202,204 | ||
| High-fat diet-induced obese female mice | Improves glucose tolerance and increases liver citrate synthase activity and triglyceride accumulation | 203 | ||
| Transverse aortic constriction-stressed mice, male conditional knockout mice Male cardiac-specific Fxn-knockout mice (Friedreich ataxia cardiomyopathy model) and male Sirt3-knockout/Fkn-knockout mice |
Improves mitochondrial function and protects mice from heart failure Improves cardiac functions and reduces energy waste and improve energy utilization in Fxn-knockout mice but not in Sirt3-knockout/Fkn-knockout mice |
205,207 | ||
| Rod-specific Namp-knockout mice, light-induced retinal dysfunction (129S1/SvlmJ) | Rescues retinal degeneration and protects the retina from light-induced injury | 206 | ||
| Alzheimer disease-relevant (AD-Tg) male and female mice | Decreases APP levels and increases mitochondrial function | 161 | ||
| Male C57BL/6 mice Male C57BL/6 male |
Improves carotid artery endothelium-dependent dilation Rescues neurovascular coupling responses by increasing endothelial NO-mediated vasodilation and improves spatial working memory and gait coordination |
208,209 | ||
| Intracerebroventricular infusion of Aβ1–42 oligomer in male Wister rats APPswe/PS1dE9 transgenic mice (Alzheimer disease model) and wild-type mice |
Improves learning and memory Improves cognitive function |
176,177 | ||
| Cerebral ischaemia in male C57BL/6 mice Collagen-induced intracerebral haemorrhage in male CD1 mice Male tissue plasminogen activator-treated cerebral ischaemia CD1 mice |
Reduces cell death of neurons and improves neurologica outcome Reduces brain oedema and cell death and promotes the recovery of body weight and neurological function Decreases mortality, brain infarction, oedema, apoptosis and haemorrhage and protects blood-brain-barrier integrity |
234–236 | ||
| Male and female Tg(SIRT2);Bubr1H/+ mice | Increases NAD+ levels and restores BubR1 levels | 237 | ||
| Nicotinamide riboside | C57BL/6 wild-type or muscle-specific Nampt-knockout (male or female) mice Wild-type or mdx mice, and Mdx/Utrn-knockout mice Male C57BL/6 muscle stem cell-specific Sirt1– knockout mice and mdx mice 70–80-year-old men |
Restores muscle mass, force generation, endurance and mitochondrial respiratory capacity in Nampt-knockout mice Improves muscle function and reduces heart disease Improves endurance, grip strength and recovery from cardiotoxin-induced muscle injury in aged mice, induces the mitochondria unfolded protein response and delayed senescence in stem cells, and increases lifespan Suppresses specific circulating inflammatory cytokine levels; no change in mitochondrial biogenesis and metabolism |
188,197, 198,238 |
|
| Male high- fat diet- fed C57BL/6 wild- type, high- fat diet- fed liver- specific Sirt1– knockout and high- fat diet- fed Apoe– knockout KK/HIJ mice Male C57BL/6 wild- type or liver- specific Nampt– knockout mice |
Prevents fatty liver and induces the mitochondrial unfolded protein response with diminished effects in Sirt1– knockout mice Improves glucose homeostasis, increases adiponectin level, and lowers hepatic cholesterol level. Improves liver regeneration, reduces hepatic steatosis and reverses the poor regeneration phenotype in liver- specific Nampt– knockout mice |
239–241 | ||
| Female Sprague Dawley rats Male high-fat diet-fed C57BL/6 wild-type mice |
Prevents or reverses paclitaxel-induced hypersensitivity to pain, no change in locomotor activity Improves glucose tolerance, reduces weight gain and hepatic, steatosis and protects against diabetic neuropathy |
242,243 | ||
| C57BL/6 wild- type or Xpa−/−/Csa−/− mice Male C57BL/6 wild- type and Atm−/− mice Male C57BL/6 wild- type and Csbm/m mice |
Lowers mitochondrial membrane potential and reactive oxygen species production Improves motor coordination and behaviour and increases the number of Purkinje cells and Atm−/− mouse Lifespan Improves the function of mitochondria isolated from cerebellum |
33,135,244 | ||
| 3xTgAD and 3xTgAD/Polb+/− mice iPS cell- derived dopaminergic neurons from patients with GBA- related PD |
Decreases phosphorylated tau levels, no effect on Aβ levels, increases neurogenesis, LTP and cognitive function, and reduces neuroinflammation (NLRP3, caspase 3) Ameliorates mitochondrial function by increasing the mitochondrial unfolded protein response (HSP60) and increases autophagy and lysosomal function |
137,175 | ||
| Cisplatin- induced acute kidney failure in C57Bl/6NTac mice | Decreases blood urea nitrogen levels and levels of markers od glomerular dysfunction | 211 | ||
| NAD+ biosynthesis modulators | P7C3 | Osteosarcoma cell line U2OS and lung cancer cell line H2122 cells | Protects cultured cells from doxorubicin-mediated toxicity by enhancing NAMPT activity | 214 |
| SBI-797812 | Lung carcinoma A549 cells and male C57BL/6J mice | Increases NMN and NAD+ levels by increasing NAMPT activity and modest increase of hepatic NAD+ levels in mice | 214,215 | |
| TES-991 and TES-102524 | MCD diet- induced non- alcoholic fatty liver disease and ischaemia–reperfusion- induced acute kidney injury in male C57BL/6J wild- type mice | Boosts de novo NAD+ synthesis by inhibiting ACMSD and increases mitochondrial functions in liver, kidney and brain | 216 | |
| CD38 inhibitors | Apigenin | High-fat diet-fed C57BL/6 wild-type mice | Decreases global protein acetylation and improves glucose and lipid homeostasis | 226 |
| Luteolinidin | Heart ischaemia-reperfusion model in male Sprague Dawley rats | Improves vascular and cardiac contractile function by restoring NADP(H) and NAD(H) pools | 228 | |
| 78c | Diet- induced obese C57Bl6 wild- type mice C57BL/6 wild-type mice Heart ischaemia–reperfusion model in male C57Bl/6J wild- type mice |
Elevates NAD+ levels in liver and muscle Improves glucose tolerance, muscle function, exercise capacity and cardiac function by mitigating mTOR– p70S6K/ERK and telomere- associated DNA damage Pathways Protects against both postischaemic endothelial and cardiac myocyte injury. Enhances preservation of contractile function and coronary flow, and decreases infarction |
91,230,231 |
Aβ, amyloid-β ; ACMSD, α-amino-β-carboxymuconate ε-semialdehyde decarboxylase; APP, amyloid precursor protein; GBA, β-glucocerebrosidase; iPS cell; induced pluripotent stem cell; LTP, long-term potentiation; MCD diet, methionine–choline-deficient diet; NAD+, nicotinamide adenine dinucleotide; NMN, nicotinamide mononucleotide; NAMPT, nicotinamide phosphoribosyltransferase; PD, Parkinson disease.
Deregulation of immune cell function
Inflammageing is now being recognized as a hallmark of ageing and a key driver of disease (FIG. 3Aa), including ageing-associated diseases, and has been described as a prominent risk factor for morbidity and death95–97. Chronic inflammation has profound effects on systemic metabolism, via a complicated crosstalk between immune cells and metabolic cells, such as hepatocytes and adipocytes, influencing metabolic processes such as glucose and lipid uptake, and insulin sensitivity96. Despite the increased interest in immunometabolism in the last decade, little is known about how NAD+ influences chronic inflammation and immune cell function. We will discuss the emerging picture next.
Innate immunity.
Chronic low-grade inflammation, characterized by aberrant activation of the innate immune system, enhanced expression of proinflammatory cytokines, such as tumour necrosis factor (TNF), IL-6 and IL-1β, and activation of immune complexes, such as the NLRP3 inflammasome, is now being recognized as a key driver of ageing-related and metabolic diseases. Additionally, it is now emerging that altered macrophage activation and phenotypic polarization is a key source of this inflammation. For example, in obese tissues such as the visceral fat, peripheral monocytes are recruited by stressed adipocytes, leading to a progressive infiltration of proinflammatory M1-like macrophages and the displacement of the resident, anti-inflammatory M2 macrophages96. This switch in macrophage polarization states in the visceral fat is accompanied by enhanced expression of proinflammatory cytokines, insulin resistance and reduced rates of lipolysis. Elie Metchnikoff, the discoverer of the macrophage, was the first to observe the increased abundance of macrophages in ageing tissues more than 100 years ago. Although this observation was initially disregarded, there is now growing evidence that ageing not only leads to increased macrophage abundance but is also accompanied by altered macrophage polarization state and function, which is emerging as a key driver of inflammageing98,99.
Some of the earliest work suggesting that NAD+ affects macrophage function showed that inhibition of NAMPT and subsequent depletion of NAD+ pools in macrophages decreased the secretion of proinflammatory cytokines, such as TNF, and caused morphological changes, such as reduced spreading in these cells100,101. Recent studies have shown that NAD+ is a critical regulator of macrophage function and that macrophage activation is associated with the upregulation of NAD+ biosynthetic or degradative pathways, depending on the acquired fate. For example, our laboratory recently showed that proinflammatory (M1) macrophage polarization is associated with enhanced expression of CD38, leading to increased NAD+ consumption45. Conversely, anti-inflammatory (M2) macrophage polarization was associated with an increase in NAD+ levels that depended on NAMPT45. Blocking the NAM salvage pathway (BOX 1) in both M1 macrophages and M2 macrophages significantly reduced gene expression of select genes associated with the M1 and M2 phenotypes. This effect could be rescued with supplementation with the NAD+ precursors NMN and NR, which bypass and rescue NAMPT inhibition45. M2 macrophages required significantly more NR/NMN to rescue macrophage activation than M1 macrophages, suggesting that NAD+ is a critical metabolite for general macrophage activation, and its metabolism is differentially regulated to control distinct biological processes and functions in M1 and M2 macrophages.
Consistent with our results, a recent study found that M1 macrophage polarization is associated with enhanced degradation of NAD+ and that inhibiting NAMPT blocks glycolytic shifts in M1 macrophages, limiting proinflammatory responses in vitro and reducing systemic inflammation in vivo in response to sepsis102. This study and another recent report6,102 suggest that this NAD+ turnover, particularly during the first few hours of macrophage M1 polarization, depends on reactive oxygen species-induced DNA damage and activation of PARP1. However, recently published results from our laboratory detected no evidence of DNA damage or PARP1 activation during M1 macropahge polarization45. By contrast, our results suggest that CD38 is the primary NAD+-consuming enzyme in M1 macrophages. These results are consistent with recent work showing that M1 macrophages are protected from reactive oxygen species-induced DNA damage as a result of increased transcription of genes involved in antioxidant defence, such as SOD2 (REFS102–104). Thus, these contradictory observations highlight a need to better delineate the major consumers of NAD+ during proinflammatory and anti-inflammatory macrophage polarization to determine whether the observed decline in NAD+ levels depends on context and time and to determine the molecular mechanisms through which NAD+ levels influence proinflammatory and anti-inflammatory gene expression and macrophage functions.
In ageing, declining NAD+ levels are associated with increased accumulation of proinflammatory M1-like resident macrophages in the liver and fat, which are characterized by increased expression of CD38 and heightened NADase activity45,105. By in vitro and in vivo methods, it was found that these CD38-overexpressing M1-like macrophages are activated directly by inflammatory cytokines secreted by senescent cells (discussed in more detail later)45,105,106. In addition, ageing macrophages are characterized by impaired de novo synthesis of NAD+, which in itself may impact macrophage function during ageing6.
With regard to inflammageing, the studies described above suggest that proinflammatory M1-like macrophages (in addition to senescent cells; see later) may be a major source of proinflammatory cytokines in ageing tissues. Ageing is associated with increased activation of the NLRP3 inflammasome45,107, which is primarily expressed by myeloid immune cells107,108, and a subsequent increase in the expression of IL-1β, which is a major cytokine implicated in promoting ageing-related disease107. Enhanced expression of proinflammatory cytokines may drive a vicious cycle of inflammageing, leading to greater inflammation, enhanced tissue and DNA damage, further activation of major consumers of NAD+, such as CD38 and PARPs, and accelerated physiological age-related decline.
Hence, targeting macrophage immunometabolism pathways, in particular those that regulate NAD+ biosynthetic or degradative pathways, could be explored as a therapeutic strategy to activate or suppress macrophage functions and regulate macrophage polarization states. This is certainly pertinent to regulating inflammageing, but could also serve to alleviate diseases driven by chronic inflammation, such as neurodegenerative diseases and autoinflammatory diseases, and also as a therapeutic strategy in cancer, where macrophage polarization can be either tumour promoting (M2) or tumour inhibitory (M1)109.
Adaptive immunity.
Like for the innate immune system, ageing is characterized by a reduced ability to build an effective adaptive immune response due to altered functions of adaptive immune cells, known as immunosenescence.
Ageing leads to an imbalance or skewing of immune cell populations, including decreased levels of naive T and B cells, loss of T cell antigen receptor diversity and an increase in levels of virtual memory T cells. The regulatory role of NAD+ and NAD+-consuming enzymes in T cell biology has been demonstrated; however, their contribution to the ageing of the adaptive immunity is largely uncharacterized. On the one hand, extracellular NAD+ has been proposed as a danger signal110 causing cell death in specific T cell subpopulations, such as regulatory T cells111. On the other hand, NAD+ seems to exhibit immunomodulatory properties, such as influencing T cell polarization112,113. However, whether NAD+ promotes a specific T cell phenotype and whether the manipulation of NAD+ metabolism with NAD+ precursors could result in similar immunomodulatory properties is still unknown.
An established hallmark of adaptive immune ageing is the expansion of the memory population of highly cytotoxic CD8+CD28− T cells114, characterized by high secretion of effector molecules such as granzyme B115. This cell population is characterized by decreased SIRT1 and FOXO1 levels, resulting in an enhanced glycolytic capacity and granzyme B production116. These studies highlight the potential of metabolic reprogramming via manipulation of NAD+-associated pathways in age-related adaptive immune dysfunction116. The upregulation of the NAD+–SIRT1–FOXO1 axis via CD38 inhibition increases effector functions in T helper 1 and 17 hybrid cells117, and future studies using CD38 inhibitors or NAD+ precursors may potentially show the therapeutic potential of targeting NAD+ in the ageing adaptive immune system.
Another immunological feature of adaptive immune ageing is the increase in the number of exhausted T cells, characterized by the expression of inhibitory receptor molecules (for example, PD1 and TIM3), decreased proliferative capacity and decreased effector functions118,119. PD1 is a component of the immune checkpoint, blockade of which is commonly used as an anticancer strategy120,121, but also has been proposed to restore the effector function of aged T cells122. With respect to NAD+ metabolism, a recent study showed that CD38 overexpression was associated with a dysfunctional and exhausted CD8 T population in PD1 blockade-resistant cancers123,124, highlighting the potential of expanding studies on CD38 inhibition to age-related exhausted T cells. However, although extremely intriguing, this hypothesis is yet to be explored in depth, and more studies will be needed to determine the preclinical efficacy of this approach.
Thus, overall, more work needs to be done to determine whether manipulating NAD+ levels is effective in reversing ageing-related immunological dysfunction in the adaptive immune system and, equally important, whether such manipulations are safe.
Cellular senescence
During ageing, cells exposed to metabolic, genotoxic or oncogene-induced stress undergo essentially irreversible cell cycle arrest known as cellular senescence. One major phenotype of senescent cells and how they are thought to promote disease is the increased expression of inflammatory mediators, mostly cytokines and chemokines, known as the senescence-associated secretory phenotype (SASP), which contributes to impaired tissue homeostasis by interfering with stem cell regeneration, tissue and wound repair and inflammageing125,126 (FIG. 3Aa). As the numbers of senescent cells gradually increase with age, cellular senescence has been linked to several age-associated diseases, and clearance of senescent cells with pharmacologic senolytics may be an effective treatment for several previously untreatable diseases, including Alzheimer disease127–129. In addition, treatments aimed at boosting cellular NAD+ levels during ageing are promising targets for extending healthspan130, but how NAD+ impacts cellular senescence is not clear. It was recently shown that senescent cells upregulate the expression of the NAM salvage enzyme NAMPT (BOX 1) and that the SASP of senescent cells depends on NAD+ levels131. Treating senescent cells with NMN can heighten the SASP, leading to increased chronic inflammation, and can promote the development of inflammation-driven cancers. These findings suggest that administration of NAD+-boosting supplements, such as NR and NMN, may come at the cost of long-term side effects, such as enhancing chronic inflammation and cancer development. Thus, a greater understanding of the benefits and unattended side effects of boosting NAD+ levels will be an important area of focus in future studies and ongoing clinical trials. As inflammation is a very complex and multipurpose process, additional studies are needed to better understand how NAD+ levels influence different inflammatory states and in what context, and to determine how, mechanistically, NAD+ metabolism impacts the biology of inflammatory immune and senescent cells.
Despite well-documented observations of senescent cells accumulating in ageing tissues and the concomitant decline of NAD+ levels in these tissues, no studies link the accumulation of inflammatory senescent cells to NAD+ levels during ageing. Recently, it was shown that CD38 levels increase in mammalian tissues with age, and CD38 has been proposed to be the major NAD+-consuming enzyme responsible for the decline in NAD+ levels during ageing42,91. However, the mechanisms driving increased CD38 expression in aged tissues and which cells express CD38 in these tissues are unclear. Recent observations suggest that innate immune cells, especially macrophages, may be the major cell population responding to the SASP with NAD+ degradation, thereby contributing to organism-wide decline in NAD+ levels45,105. We showed that macrophages co-cultured in or exposed to conditioned media from senescent cells have enhanced expression of the NAD+-consuming enzyme CD38 and increased proliferation45. Importantly, another group has also independently shown that senescent cells and their SASP activate CD38 expression and promote CD38-dependent NADase activity in macrophages105,106. Furthermore, we showed in ageing mouse models and mice treated with the senescence-inducing chemotherapeutic reagent doxorubicin that accumulation of senescent cells in metabolic tissues, such as the visceral fat and liver, can directly activate the expression of CD38 in tissue-resident macrophages45. In further support of the connection between inflammageing, senescent cell burden and NAD+, a recent study in mice has also shown that cells with dysfunctional mitochondria initiate a proinflammatory programme, with the secretion of proinflammatory cytokines. This was associated with greater senescent cell burden, increased metabolic and physical dysfunction, and premature ageing132. In this context, supplementation with NR was able to partially rescue this multimorbidity syndrome, in part by reducing inflammation and senescent cell burden132 — in contrast to the finding discussed above that NAD+ precursors can increase SASP expression131. Thus, regulation of senescence by NAD+ appears to be complex.
Overall, these findings suggest that targeting immune cells such as T cells, macrophages and senescent cells should be considered in approaches aiming to restore NAD+ levels during ageing. However, until more is known about the long-term side effects of enhancing NAD+ levels, this work should proceed with caution.
Neurodegeneration
Ageing is strongly associated with most neurodegenerative diseases and is accompanied by decreased cellular NAD+ levels in the mammalian brain133. NAD+ depletion is reported in several models of accelerated ageing which exhibit neurodegeneration33,134,135, as well as in neuro-degenerative diseases, including Alzheimer disease136, Parkinson disease137,138 and amyotrophic lateral sclerosis (ALS)139. The underlying cause and mechanisms of NAD+ loss in the brain during age-related neurodegenerative diseases are still largely unknown. However, multiple lines of evidence support a neuroprotective role for NAD+ (FIG. 3Ab).
First, axonal degeneration, which is a precursor to many age-related neuronal disorders140,141, is characterized by rapid NAD+ depletion. During normal physiological conditions, the NAD+ biosynthetic enzyme nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) (FIG. 1; BOX 1) is a survival factor in axons, in which it needs to be constantly replenished via anterograde axonal transport due to its rapid turnover142. However, during axon degeneration, NMNAT2 axonal transport is blocked, and the axonal pool of the protein is rapidly degraded, leading to a critical depletion of NAD+ in the axon142,143. Moreover, the NAD+-consuming enzyme SARM1 is activated by axonal injury and mediates axonal degeneration by promoting NAD+ degradation60. This was originally shown in Wallerian degeneration slow (Wlds) mice, which are protected from axonal degeneration, showing absence of SARM1 expression144 and higher neuronal NAD+ levels, owing to the overexpression of a chimeric fusion protein of NMNAT1, which allows its redistribution from the nucleus to the axon, where it can substitute for the activity of NMNAT2 (REFS145,146). The role of SARM1 in promoting axonal damage has been demonstrated in several other in vivo models. Sarm1-knockout mice are protected from axonal degeneration60 and can rescue the severe axon growth defects and perinatal death induced by lack of NMNAT2 (REFS147,148). Recently, a transgenic mouse model overexpressing a dominant negative version of SARM1 in neurons showed a significant delay in axon degeneration and hints at the promising effect of gene therapy or small molecules targeting SARM1 for treating neuropathies149.
Overall, Wlds mouse studies largely show the neuro protective function of the biosynthetic enzymes NMNAT1, NMNAT2 and NMNAT3 (REFS142,150–152) and their protective role in several neurodegenerative disorders, including Parkinson disease153–155. However, the mechanism remains unclear. Besides the regulation of SARM1-dependent NAD+ degradation discussed above, the degradation of their substrate and NAD+ precursor, NMN, by NMNATs seems to protect axons from degeneration. In contrast to the neuroprotective role of NAD+, the precursor NMN has been reported to have a neurotoxic role promoting SARM1 activation and cyclic ADP-ribose generation, leading to axonal destruction156. However, the evidence that NMN accumulation leads to axonal degeneration needs to be validated and remains controversial. Nonetheless, this possibility raises questions about the therapeutic potential of increasing NAD+ synthesis by supplementing NAD+ precursors such as NMN.
Additional lines of evidence supporting a neuroprotective role for NAD+ also include studies using P7C3, an aminopropyl carbazole reported to be an allosteric activator of NAMPT in the NAM salvage pathway (BOX 1). P7C3 was shown to be neuroprotective in mouse models of Parkinson disease137, Alzheimer disease157 and ALS158. Furthermore, NAD+-consuming enzymes other than SARM1 have been reported to play a role in intracellular NAD+ depletion during ageing-related neurodegenerative diseases. For example, CD38 expression increases in the course of Alzheimer disease progression159–161, and an Alzheimer disease mouse model lacking CD38 (Cd38-knockout mice), which results in elevated NAD+ levels in their brains, shows a milder disease phenotype159. Consistent with the neuroprotective role of NAD+, Cd38-knockout mice are also protected from neuronal cell death on ischaemic brain injury162. Multiple cells in the brain, including microglia, astrocytes, neurons and endothelial cells, express CD38 (REFS108,160), and treating microglia and astrocytes with inflammatory cytokines induces CD38 expression163,164. Consistent with this finding, CD38 expression was associated with neuroinflammation with greater amounts of proinflammatory macrophages/microglia in mouse brains160,165. CD38 was also reported to affect social behaviour, as was its homologue CD157 (REFS166,167), substantiating the functional impact of NAD+ on neuronal function. Although there is no direct evidence of a causal role of CD38 in neurodegenerative diseases, as discussed above, CD38 is emerging as a key enzyme involved in inflammageing and senescence45,105,106, which are strongly associated with neurodegenerative diseases124,128,168,169. Lastly, PARP1 activation is also associated with Alzheimer disease170,171 and Parkinson disease172,173 pathogenesis. In vivo studies using various Alzheimer disease and Parkinson disease models have shown that loss of PARP1 protects from brain dysfunctions and cognitive decline170–174. However, the contribution of PARP1 activation to NAD+ depletion during neurodegeneration remains largely unexplored.
From the findings taken together, there is growing evidence that NAD+ is a central metabolite for maintaining a healthy nervous system and can impact the biology of multiple brain cell types, suggesting that counteracting the ageing-associated decline in NAD+ levels may be a viable therapeutic approach to treat neurodegenerative diseases. Restoration of NAD+ levels with NAD+ supplements and the overexpression of the two biosynthetic enzymes NAMPT and NMNAT1 were found to prevent axon degeneration60,146. Moreover, the NAD+ precursors NR and NMN improve neuronal cell health, memory and cognitive function in rat and mouse models of Alzheimer disease136,161,175–180, and also showed neuroprotective properties in Drosophila melanogaster models of Parkinson disease181,182 and in mouse models of ALS139. Importantly, there are now several clinical trials in progress using NAD+ precursors, especially NR, to treat neurological disorders68 and to promote healthy ageing (TABLE 2). These trials will undoubtedly expand our understanding of NAD+ metabolism in the human neurodegenerative processes68.
Table 2 |
Human clinical trials focusing on ageing
| NAD+ precursor | Description | Design | Dose and duration | NCT/UMIN no. |
|---|---|---|---|---|
| NMN | Study of efficacy against insulin sensitivity and β- cell functions in elderly women | Randomized, placebo-controlled, double-blind study Postmenopausal and prediabetic women Age 55–75 years |
Oral administration Long-term NMN administration: 250 mg daily for 8 weeks |
{“type”:”clinical-trial”,”attrs”:{“text”:”NCT03151239″,”term_id”:”NCT03151239″}}NCT03151239 |
| Study of pharmacokinetics and safety in healthy volunteer | Non-randomized, open-label, non-placebo-controlled study Male healthy volunteers Age from 40 to 60 years |
Oral administration Single administration of 100, 250 or 500 mg NMN |
UMIN000021309 | |
| Study of pharmacokinetics, safety and effects with regard to various hormonal levels in healthy volunteers | Randomized, dose-comparison, double-blinded study Healthy volunteers Age from 50 to 70 years |
Oral administration Long-term NMN administration: 100 mg or 200 mg for 24 weeks |
UMIN000025739 | |
| Study of pharmacokinetics, safety and efficacy with regard to glucose metabolism in healthy volunteers | Non-randomized, open-label, non-placebo controlled study Male healthy volunteers Age from 40 to 60 years |
Oral administration Long-term NMN administration for 8 weeks. Dose is not described |
UMIN000030609 | |
| Study of pharmacokinetics, safety and efficacy with regard to blood pressure and physical endurance in healthy volunteers | Multicentre, randomized, double-blind, placebo-controlled study Healthy volunteers Age from 40 to 65 years |
Oral administration Long term NMN administration; 300 mg daily for 60 days |
{“type”:”clinical-trial”,”attrs”:{“text”:”NCT04228640″,”term_id”:”NCT04228640″}}NCT04228640 | |
| Nicotinamide riboside | Study of safety and efficacy with regard to physical activities in elderly people | Non-randomized, open-label, crossover study Healthy volunteers Age from 55 to 79 years |
Oral administration Crossover of placebo for 6 weeks and NR 500 mg twice daily for 6 weeks |
{“type”:”clinical-trial”,”attrs”:{“text”:”NCT02921659″,”term_id”:”NCT02921659″}}NCT02921659 |
| Study of efficacy with regard to bone, skeletal muscle and metabolic functions in ageing | Randomized, double-blind, placebo-controlled study Female healthy volunteers Age from 65 to 80 years |
Oral administration 1,000 mg NR daily in a regimen of 500 mg every 12 hours for 4.5 months After 4.5 months, advanced individual training will be implemented with administration of 1,000 mg daily (500 mg every 12 hours) for a further 6 weeks. Total of 6 months |
{“type”:”clinical-trial”,”attrs”:{“text”:”NCT03818802″,”term_id”:”NCT03818802″}}NCT03818802 | |
| Study of efficacy with regard to elevated systolic blood pressure and arterial stiffness in middle-aged and older adults | Randomized, placebo-controlled, double-blind study Healthy volunteers Age from 50 to 79 years |
Oral administration Long-term NR administration: 1,000 mg daily for 3 months |
{“type”:”clinical-trial”,”attrs”:{“text”:”NCT03821623″,”term_id”:”NCT03821623″}}NCT03821623 | |
| Study of recovery phase and improved outcome after acute illness | Randomized study Hospitalized patients with tissue damage Age from 18 years |
Oral administration Long-term NR administration: 250, 500 or 1,000 mg daily for 3 months |
{“type”:”clinical-trial”,”attrs”:{“text”:”NCT04110028″,”term_id”:”NCT04110028″}}NCT04110028 | |
| Study of pharmacokinetics and efficacy with regard to brain function, including cognition and blood flow in people with MCI | Randomized study Patients with MCI Age from 65 years |
Oral administration NR dosage increased from 250 mg daily to 1 g daily as tolerated |
{“type”:”clinical-trial”,”attrs”:{“text”:”NCT02942888″,”term_id”:”NCT02942888″}}NCT02942888 | |
| Study of cognitive performance in subjective cognitive decline and mild cognitive impairment in ageing | Crossover, randomized block sequence, double-blind, placebo-controlled study Patients with MCI Age from 60 years |
Oral administration Long-term NR administration: 1,200 mg for 8 weeks |
{“type”:”clinical-trial”,”attrs”:{“text”:”NCT04078178″,”term_id”:”NCT04078178″}}NCT04078178 | |
| Study of safety and efficacy with regard to NAD+ sustainability in elderly people | Randomized, placebo-controlled, double-blind study Healthy volunteers Age from 60 to 80 years |
Oral administration 1XNRPT (250 mg NR and 50 mg pterostilbene) once daily or 2XNRPT (500 mg NR and 100 mg pterostilbene) twice daily for 8 weeks |
{“type”:”clinical-trial”,”attrs”:{“text”:”NCT02678611″,”term_id”:”NCT02678611″}}NCT02678611 | |
| NAM | Study of the efficacy a mixture of the NAD+ precursors NA, NAM and tryptophan with regard to physical function and skeletal muscle mitochondrial function in physically compromised, elderly humans | Randomized, double-blind, crossover trial Healthy volunteers Age from 65 to 75 years |
Oral administration Long-term administration of NAD+ precursors: total of 204 NE per serving in a whey protein source for 31 days |
{“type”:”clinical-trial”,”attrs”:{“text”:”NCT03310034″,”term_id”:”NCT03310034″}}NCT03310034 |
| Exploratory study on phosphorylated tau protein changes in CSF in mild cognitive impairment due to Alzheimer disease and mild Alzheimer disease dementia | Double-blind, randomized, placebo-controlled study Patients with Alzheimer disease Age from 50 years |
Oral administration Long-term administration of NAM: 1,500 mg daily for 48 weeks |
{“type”:”clinical-trial”,”attrs”:{“text”:”NCT03061474″,”term_id”:”NCT03061474″}}NCT03061474 | |
| Study of efficacy with regard to the severity of Parkinson disease symptoms | Double-blind, randomized study Patients with Parkinson disease Age from 35 years |
Oral administration Long-term administration of NAM: 200 mg daily for 18 months |
{“type”:”clinical-trial”,”attrs”:{“text”:”NCT03808961″,”term_id”:”NCT03808961″}}NCT03808961 |
CSF, cerebrospinal fluid; MCI, mild cognitive impairment; NA, nicotinic acid; NAD+, nicotinamide adenine dinucleotide; NAM, nicotinamide; NE, niacin equivalents; NMN, nicotinamide mononucleotide; NR, nicotinamide riboside.
Therapeutic targeting of NAD+ level decline
Over the past two decades, the importance of NAD+ in healthy ageing and longevity has been recognized. Preclinical studies in different animal models, such as Caenorhabditis elegans, D. melanogaster, rodents and human primary cells, have firmly established that there is an age-dependent decline of NAD+ levels, which ranges from 10% to 65%, depending on different organs and age130. NAD+ can be regulated by dietary and lifestyle choices (FIG. 4). It can also be modulated pharmacologically, and so far, three main approaches have been explored to increase NAD+ levels: dietary supplementation with NAD+ precursors involved in the salvage pathways of NAD+; modulation of NAD+ biosynthetic enzymes, in particular those that regulate the rate-limiting step of de novo synthesis and salvage pathways (α-amino-β-carboxymuconate ε-semialdehyde decarboxylase (ACMSD) and NAMPT, respectively); and the inhibition of enzymes involved in NAD+ degradation, such as PARPs and CD38. Augmentation of NAD+ levels has shown efficacy in a variety of mouse models of human diseases (TABLE 1), leading to numerous clinical trials of NAD+ boosters in humans during ageing (TABLE 2). Considering the scope of this Review, we will not detail the NAD+-related clinical trials and we refer the reader to more comprehensive reviews of this topic68,183,184.
Most of the preclinical studies in rodents suggest a strong translational potential for NAD+-boosting therapies. Studies in humans are less advanced, and to date the clinical trials evaluating the pharmacokinetics and toxicology of NAD+ precursors have primarily demonstrated that NMN and NR administration is safe and can efficiently increase NAD+ levels in healthy volunteers (TABLE 2). Of note, there have been more phase I clinical trials using NR than those using NMN, and these have provided conflicting results. One trial showed that short-term NR administration has some beneficial effects in healthy elderly individuals185, and another showed positive results in patients with ALS93,186,187. However, NR has shown little to no effect in aged men with obesity93,186,188 (see also the discussion in the section Metabolic dysfunction and TABLE 2). Therefore, further human clinical studies are needed to determine the proper dose, treatment period and long-term toxicological outcome, together with consideration of the diversity of participants, to better address the translation of the NAD+-boosting strategy.
Dietary supplementation
The effects of different NAD+ precursors on the lifespan and healthspan of yeast and C. elegans have been extensively investigated. In both wild-type yeast and C. elegans, low (micromolar) concentrations of NR extend lifespan in a sirtuin-dependent manner135,189,190. In addition, lifespan extension was observed on administration of NAM to wild-type C. elegans190. However, a supraphysiologic dose of NAM (1–5 mM as compared with 200 μM used in experiments with lifespan extension190) has also been associated with reduced lifespan in yeast and C. elegans190–192. Thus, it is unclear whether NAM administration is beneficial or detrimental in the context of ageing. One potential explanation for this discrepancy is that NAM influences NAD+ functions directly and indirectly. Aside from being a direct NAD+ precursor in the salvage pathway, NAM is also a by-product of NAD+ catabolism, and at high millimolar concentrations it has been shown to act as a feedback inhibitor for NAD+-dependent enzymes, such as PARPs193 and sirtuins194 in vitro. However, the results regarding the inhibitory effects of a high dose of NAM were inconclusive in several animal studies195. Another possible reason for the inhibitory role of NAM on NAD+ activity could be that an increase in NAM level leads to a proportional increase of NAM methylation. This in turn affects cellular availability of methyl groups, which are important for DNA methylation and gene expression modulation (BOX 2). High levels of methylated NAM have been associated with the pathogenesis of type 2 diabetes, Parkinson disease and cardiac diseases195. Therefore, further studies are needed to elucidate the safest NAM doses and treatment periods.
The strategy to boost NAD+ has also been adopted in extending lifespan in age-related disorders. NAD+ replenishment, using NR and/or NMN, has also been effective in delaying progeroid phenotype and extending lifespan in relevant C. elegans models of xeroderma pigmentosum group A, ataxia telangiectasia and Cockayne syndrome33,134,135 and in a worm model of Werner syndrome (a progeroid disease)196. NMN treatment significantly increased NAD+ levels and the maximum lifespan also in a mouse model of ataxia telangiectasia135. Treatment with NR also improved muscle stem cell functions by regulating mitochondrial metabolism in a mouse model of muscular dystrophy197,198.
Few studies to date have investigated the effects of NMN, NAM and NR on prolonging healthspan and/or lifespan in wild-type mice. In one study, long-term (1-year) feeding of mice with NMN starting at 5 months of age increased whole-body insulin sensitivity, energy metabolism and physical activity and improved lipid profiles with no obvious toxic effects199. In another study, long-term administration of NAM prevented disease related to a high-fat diet, promoting healthspan, but it did not have significant effects on lifespan200. Finally, NR administration in old mice showed a small but significant increase in lifespan (5%)198. Reflecting these results, most of the preclinical studies using NAD+ precursors have focused on extending the healthspan by counteracting different age-related diseases characterized by declines in NAD+ levels130 (TABLE 1). It has been demonstrated that NMN enhances mitochondrial function in various organs, including skeletal muscle201, kidney202–204, liver203, heart205, eyes206, brain161 and the cardiovascular system207, and can mitigate oxidative stress in the vasculature208, including improvements in neurovascular coupling responses in the aged cortex of the brain209. These effects of NMN supplementation may be mediated by sirtuins. For example, SIRT3 mediates NMN-induced improvements in cardiac and extracardiac metabolic function in a mouse model of cardiomyopathy207. NMN supplementation also increased the number of endothelial cells and improved endothelial cell function by increasing blood flow and endurance in elderly mice by a SIRT1-dependent mechanism210.
Lastly, it has been shown that a reduced form of NR, NRH, is more stable and potent than NR in increasing intracellular NAD+ content, which occurs through a new metabolic pathway in which adenosine kinase converts NRH into NMNH, which is then oxidized to NMN or further metabolized by NMNATs to produce NADH and then NAD+ (REFS211,212). Thus, future studies will be needed to compare the efficacy of using NRH relative to the more commonly used NAD+ precursors NAM, NR and NMN as a therapeutic, along with its potential side effects.
In summary, the promising preclinical results discussed above for NAD+ precursors demonstrate that NAD+ augmentation may promote healthspan and even lifespan in mammals. These beneficial effects of NAD+ precursors could possibly be relevant to humans and are currently being tested in clinical trials (TABLE 2).
Modulation of NAD+ biosynthesis
The salvage and de novo synthesis pathways of NAD+ are also potential targets for therapies boosting NAD+ levels in vivo. Specifically, activators of NAMPT, the rate-limiting enzyme in the NAD+ salvage pathway, and NMNATs, which contribute to the NAD+ de novo and salvage pathways (FIG. 1), have been suggested as possible therapeutic interventions for boosting tissue NAD+ levels213. Indeed, the neuroprotective agent P7C3 enhances NAMPT activity and boosts NAD+ levels in doxorubicin-treated human cells, suggesting it may be a functional therapeutic for ageing and age-related disease processes, such as neurodegeneration214. However, the real efficacy of P7C3 in increasing NAMPT activity remains controversial215. Recently, another small molecule, SBI-797812, was proposed as a potent NAMPT activator active in the nanomolar range. This compound increased NAMPT-mediated NMN production in vitro and, importantly, NAD+ levels in cell lines and in vivo214,215. Despite the limited effect of SBI-797812 of boosting only hepatic NAD+ levels in vivo, this is a promising pharmacological approach to raise intracellular NAD+ levels. Moreover, pharmacological inhibition of ACMSD by TES-991 and TES-102524 boosts de novo NAD+ synthesis and SIRT1 activity, ultimately enhancing mitochondrial function in the liver, kidneys and brain of mice216. Although these strategies are promising for therapeutic increase of NAD+ levels, one of the challenges that remains is identifying with greater precision how some of these molecules, such as P7C3, work at the molecular level, determining their targets and exploring potential off-target effects.
In addition to modulating NAD+ levels, in recent years, attention has been given to modulating the NAD+/NADH redox balance primarily through the administration of the redox-cycling quinone β-lapachone, an exogenous co-substrate of NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), which regenerates NAD+ from NADH. NQO1 was shown to be increased by caloric restriction (although it was not sufficient to mediate anti-ageing effects of this dietary intervention)217, and enhancing NQO1 activity by administering β-lapachone prevented age-dependent decline of motor and cognitive function in aged mice by ameliorating mitochondrial dysfunction218,219. However, NQO1 is commonly overexpressed in most solid tumours in which β-lapachone administration induces imbalance of redox cycle and oxidative stresses220; therefore, β-lapachone can have a variety of effects depending on the context and should be administered with caution.
Inhibition of NAD+ consumption
Targeting NAD+ degradation enzymes and pathways is perhaps the most compelling area of recent therapeutic development. Specifically, targeting PARPs and NADases, including CD38, CD157 and SARMs, has great potential to treat age-related diseases associated with declines in NAD+ levels. In C. elegans, PARP inhibition results in a significant lifespan extension in wild-type worms190, in a model of ataxia telangiectasia135 and under hyperglycaemic conditions221.
PARP1 inhibitors, such as olaparib and rucaparib, are marketed as adjuncts to chemotherapy or monotherapies for cancer222. They sensitize tumours to DNA damage, but they have limited use due to toxicity223–225. CD38 is one of the main NADases in mammals and plays a key role in age-related decline of NAD+ levels (see the section NAD+-dependent mechanisms in ageing)42. Several inhibitors of CD38 already exist or are in development, a few of which enhance NAD+ levels in vivo. For example, apigenin, a naturally occurring flavonoid, enhances NAD+ levels in human cells and mouse liver tissue, and improves glucose and lipid homeostasis in mouse models of obesity226. More recently, apigenin has been shown to downregulate CD38 expression, and to increase the intracellular NAD+/NADH ratio and SIRT3-mediated mitochondrial antioxidative enzyme activity in the kidneys of diabetic rats227. Another flavonoid CD38 inhibitor, luteolinidin, boosts NAD+ levels and protects the endothelium and myocardium after myocardial ischaemia in mice228. Clinically, luteolin has neuroprotective effects in children with autism229, although NAD+ levels were not directly assessed in this context. Additionally, several derivatives of 4-aminoquinoline, including the compound 78c, inhibit CD38 and boost NAD+ levels in mouse muscle, liver and heart230. Compound 78c also prevents age-related declines in NAD+ levels in mice, and treatment of aged mice with this compound was shown to ameliorate metabolic dysfunction, reduce DNA damage accumulation and improve muscle function91,231. Although the study authors did not report lifespan changes directly, they did report activation of the prolongevity AMPK pathway and reduced activation of the mTOR–p70S6K and ERK pathways91. More recently, compound 78c protected against postischaemic endothelial and cardiac myocyte injury in mice91,231. Overall, mounting evidence suggests that targeting CD38 and related NAD+-consuming enzymes, such as PARPs, has great potential as an NAD+-boosting target to extend human healthspan, and several pharmacological approaches are in development to inhibit CD38 and its NADase activity232 (TABLE 1).
Conclusions and perspective
Almost 90 years after its initial discovery, NAD+ is emerging as a central metabolite in the ageing field, and NAD+ level decline is becoming an established feature of several age-associated diseases. The NAD+ field is moving rapidly and has evolved as one of the major exciting research areas in biomedical research. In the past 5 years there has been a plethora of advancements, including the development of exciting tools and technology (Supplementary Box 4), to further advance our understanding of how NAD+ levels influence or are influenced by complex signalling, metabolic and cellular pathways. Furthermore, with the use of stable-isotope tracing and NAD+ biosensors, there have also been major advances and refinement in our understanding of how NAD+ levels are regulated both at the cellular level and at the systems level. We now have greater understanding of the mechanisms that lead to decline of NAD+ levels with age and the emerging role of NAD+-consuming enzymes such as CD38 and SARM1, along with PARPs in this process. Furthermore, this decline in NAD+ levels is now known to affect multiple age-dependent cellular processes, including DNA repair, oxidative stress and immune cell function. Moreover, recent pre-clinical trials have demonstrated, using different animal models, that NAD+ depletion is a key pathway in age-associated diseases, including neurodegenerative, metabolic and progeroid disorders. However, it is still not clear which cells and enzymes drive the decline in NAD+ levels in specific disorders. Moreover, which targets or pathways can be exploited to efficiently and safely restore NAD+ homeostasis is still under investigation. The good news is that different NAD+-boosting strategies have been shown to be effective in extending healthspan and lifespan (see the section Therapeutic targeting of NAD+ level decline and TABLE 1). Thus, the use of NAD+ precursors such as NMN and NR, along with small molecules that boost NAD+ biosynthesis or inhibit NAD+ degradation, offers an exciting therapeutic approach to treat ageing-related diseases and increase human healthspan. This is particularly important as elderly populations are rising rapidly, and ageing-related diseases are predicted to cause a great deal of societal and economic burden in the coming decades. However, the translatability of NAD+-boosting therapies to humans remains the key question to be answered. Several human clinical trials are ongoing to assess the safety and efficacy of NAD+ augmentation (TABLE 2) and, importantly, early-phase trials of short-term NR/NMN administration have proven it to be safe and to increase NAD+ levels in healthy participants. However, despite the promising preliminary results, whether long-term supplementation with NAD+ precursors has any side effects is still unknown. Moreover, many other questions need to be answered to deepen our understanding of the potential of NAD+-boosting therapies: is there any tissue/disease specificity for NMN and NR? What are the therapeutic doses of NAD+ precursors necessary for different diseases? Could the combination of NAD+-boosting strategies, such as CD38 and/or PARP1 inhibitors with NAD+ precursor supplementation, be something to consider? Hopefully, the upcoming results of current clinical trials will shed some light on our unsolved questions and set the basis for future directions in deciphering the role of NAD+ during ageing in humans.
Supplementary Material
Supplementary materials
Acknowledgements
This Review was supported by NIH grant R24DK085610 (E.V.) and Buck Institute for Research on Aging intramural funds (E.V.). A.J.C. is a recipient of a University of California President’s Postdoctoral Fellowship at the University of California, San Francisco, and is also supported by an NIH T32 training grant (3T32AG000266-19S1).
Glossary
| Redox reactions | Oxidation–reduction chemical reactions that involve a transfer of electrons between two species. |
| Hydride | Formally, the anion of hydrogen (H−). The term is commonly used to describe a binary compound that hydrogen forms with other electropositive elements. |
| Kynurenic acid | A quinoline-2-carboxylic acid with a hydroxy group at C-4 (4-hydroxyquinoline-2-carboxylic acid). it is a product of the metabolism of l-tryptophan. |
| Picolinic acid | A pyridinemonocarboxylic acid in which the carboxy group is located at position 2 (pyridine-2-carboxylic acid). it is an intermediate in the metabolism of l-tryptophan. |
| Extracellular vesicles | Lipid bilayer-delimited particles of endosomal and plasma membrane origin that are released by cells in the extracellular milieu. |
| Insulin resistance | An impaired response to exogenous or endogenous insulin to increase glucose uptake and utilization, resulting in elevated levels of glucose in the blood |
| Xeroderma pigmentosum | A rare autosomal recessive genetic disorder characterized by a defect in the DNA repair system (primarily in the nucleotide excision repair system) that causes increased sensitivity to the DNA-damaging effects of ultraviolet radiation. |
| Progeroid diseases | A group of rare genetic disorders characterized by clinical features typical of physiological ageing and mostly due to defects in the DNA repair system or in lamin A. |
| Ataxia telangiectasia | A rare autosomal recessive genetic disorder caused by defects in the ATM gene, which is involved in cell division and DNA repair. it is characterized by neurodegeneration, immunodeficiency, increased radiation sensitivity and cancer susceptibility. |
| Cockayne syndrome | A rare autosomal recessive genetic disorder caused by defects in the ERCC6 or ERCC8 gene, which is involved in DNA repair. it is characterized by severe photosensitivity, neurodegeneration and premature ageing. |
| Hormesis | A biphasic dose response to an agent characterized by a stimulatory or beneficial effect at low dose and an inhibitory or toxic effect at high dose. |
| AKT | A serine/threonine-specific protein kinase that participates in multiple signalling pathways related to metabolism, cell survival, motility, transcription and cell cycle progression. |
| Km | Michaelis–Menten constant representing the substrate concentration at which the reaction is half the maximum velocity (Vmax) in the Michaelis–Menten enzymatic kinetic model. |
| Vmax | The maximum reaction rate achieved by the system at saturated substrate concentration in the Michaelis–Menten enzymatic kinetic model. |
| Ectoenzymes | Enzymes located on the outer surface of a cell’s membrane with their catalytic site available to the exterior environment of the cell. |
| Transmembrane protein with a type II orientation | Integral cell membrane protein with the amino terminus on the cytoplasmic side and the carboxy terminus on the extracellular side of the membrane. The transmembrane domain is located close to the amino terminus. Type III transmembrane proteins show an opposite orientation. |
| Glycophosphatidylinositol-anchored protein | Soluble protein attached by a glycolipid anchor (glycophosphatidylinositol) to the cell membrane. |
| Paneth cells | Highly specialized epithelial cells located in the small intestine secreting antimicrobial peptides and proteins. |
| Inflammageing | Defined as the low-grade chronic inflammation and immune cell dysregulation/exhaustion that occurs gradually during the ageing process. inflammageing is emerging as a key causal factor for many age-related diseases. |
| NLRP3 inflammasome | A multiprotein complex made up of the proteins ASC, NLRP3 and caspase 1 that is activated in response to pathogens or sterile cell and tissue damage. When activated, the complex leads to the cleavage of the proforms of the cytokines IL-1β and IL-18, which themselves become activated and secreted to further amplify inflammation and immune responses. |
| Virtual memory T cells | A subpopulation of CD8+ T cells that have a memory-like phenotype (semidifferentiated) but are never been exposed to a foreign antigen (antigen naive). |
| Regulatory T cells | A subpopulation of CD4+ T cells that modulate the immune system, suppressing the immune response and maintaining tolerance to self-antigens. |
| Immune checkpoint | Molecules present on the surface of different cell types (that is, T cells, antigen-presenting cells and cancer cells) that regulate the immune response via inhibitory or activating immune signalling pathways. |
| Senolytics | Agents that target and eliminate senescent cells. |
| Wallerian degeneration | An active process of degeneration that occurs after any lesion or interruption of axons of neurons that ultimately leads to cell death. |
| Microglia | Type of glial cell located throughout the brain and spinal cord that functions as a resident macrophage and is the first and main form of active immune defence in the central nervous system. |
| Astrocytes | Highly specialized star-shaped glial cells located in the brain and spinal cord and involved in several processes, including support of the blood–brain barrier, provision of nutrients to neurons, repair and scarring following injury, and facilitation of neurotransmission |
| α-Amino-β-carboxymuconate ε-semialdehyde decarboxylase | In de novo NAD+ biosynthesis, it is the enzyme that catalyses the decarboxylation of α-amino-β-carboxymuconate ε-semialdehyde to α-aminomuconate ε-semialdehyde. |
| Flavonoid | A member of a class of polyphenolic secondary metabolites found in plants. |
| AMPK | 5′-AMP-activated protein kinase, an enzyme that on changes in the ATP:AMP ratio phosphorylates downstream targets to redirect metabolism towards increased catabolism and decreased anabolism. |
| mTOR–p70S6K | Signalling pathway that involves the serine/threonine protein kinase mTOR and the mitogen-activated serine/threonine protein kinase p70S6K and regulates protein synthesis and cell proliferation, differentiation and survival. |
| ERK | Extracellular signal-regulated kinase involved in cell growth by controlling many proteins required in translation regulation. |
Footnotes
Competing interests
E.V. is a scientific co-founder of Napa Therapeutics and serves on the scientific advisory board of Seneque. E.V., A.J.C. and R.P. receive research support from Napa Therapeutics. E.V. and A.G. receive research support from BaReCia. A.G serves as Chief Scientific Officer for Seneque USA and is one of the inventors on a patent (PCT/US18/46233) for the SLC12A8 nicotinamide mononucleotide transporter, whose applicant is Washington University in St. Louis and which has been licensed by Teijin Limited (Japan).
Peer review information
Nature Reviews Molecular Cell Biology thanks J. Auwerx and M. Hirschey and T. Nakagawa for their contribution to the peer review of this work.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s41580-020-00313-x.
Content retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7963035/.