Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling
Reactive oxygen species (ROS) are generated during mitochondrial oxidative metabolism as well as in cellular response to xenobiotics, cytokines, and bacterial invasion. Oxidative stress refers to the imbalance due to excess ROS or oxidants over the capability of the cell to mount an effective antioxidant response. Oxidative stress results in macromolecular damage and is implicated in various disease states such as atherosclerosis, diabetes, cancer, neurodegeneration, and aging. Paradoxically, accumulating evidence indicates that ROS also serve as critical signaling molecules in cell proliferation and survival. While there is a large body of research demonstrating the general effect of oxidative stress on signaling pathways, less is known about the initial and direct regulation of signaling molecules by ROS, or what we term the “oxidative interface.” Cellular ROS sensing and metabolism are tightly regulated by a variety of proteins involved in the redox (reduction/oxidation) mechanism. This review focuses on the molecular mechanisms through which ROS directly interact with critical signaling molecules to initiate signaling in a broad variety of cellular processes, such as proliferation and survival (MAP kinases, PI3 kinase, PTEN, and protein tyrosine phosphatases), ROS homeostasis and antioxidant gene regulation (thioredoxin, peroxiredoxin, Ref-1, and Nrf-2), mitochondrial oxidative stress, apoptosis, and aging (p66Shc), iron homeostasis through iron–sulfur cluster proteins (IRE–IRP), and ATM-regulated DNA damage response.
1. Introduction
Reactive oxygen species (ROS), such as superoxide anion (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (HO•), consist of radical and non-radical oxygen species formed by the partial reduction of oxygen. Cellular ROS are generated endogenously as in the process of mitochondrial oxidative phosphorylation, or they may arise from interactions with exogenous sources such as xenobiotic compounds. When ROS overwhelm the cellular antioxidant defense system, whether through an increase in ROS levels or a decrease in the cellular antioxidant capacity, oxidative stress occurs. Oxidative stress results in direct or indirect ROS-mediated damage of nucleic acids, proteins, and lipids, and has been implicated in carcinogenesis [1], neurodegeneration [2,3], atherosclerosis, diabetes [4], and aging [5]. However, ROS involvement in the pathogenesis of disease states is not confined to macromolecular damage. There is increasing evidence that ROS signaling contributes to disease. For example, ROS have been shown to promote tumor metastasis through gene activation [6]. While there exists ample evidence demonstrating the role of ROS in regulating cellular signaling pathways, the question that is raised is exactly how do ROS initiate cellular signaling? The “oxidative interface” is that boundary between ROS and the signaling molecules they activate; that is, the figurative region that describes how ROS directly activate oxidative stress-responsive pathways. This review seeks to explore the oxidative interface between ROS and a functionally broad selection of cellular signaling pathways regulating a variety of cellular processes (Fig. 1).
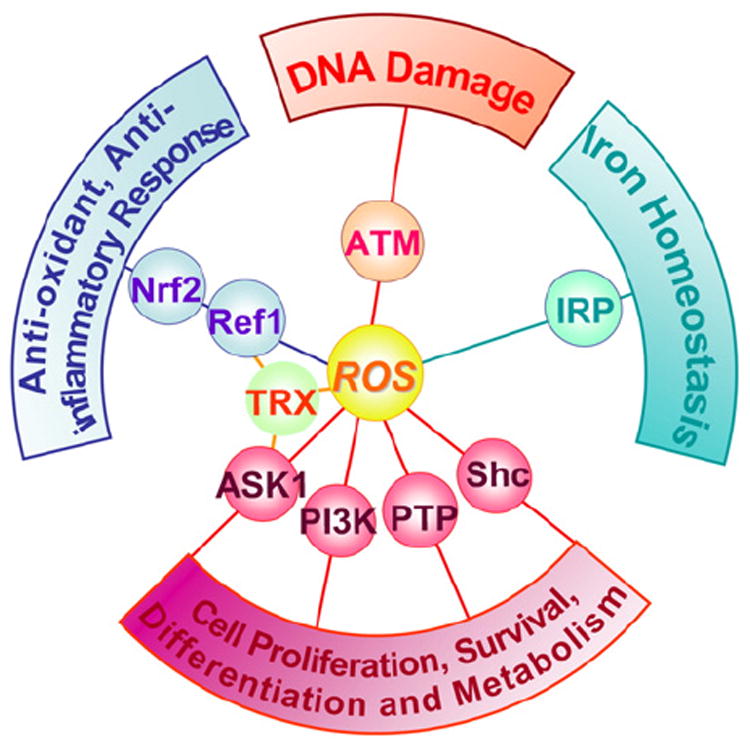
Cellular signaling pathways regulated by ROS. Reactive oxygen species (ROS) regulate several signaling pathways through interaction with critical signalingmolecules, affecting a variety of cellular processes, such as proliferation, metabolism, differentiation, and survival (apoptosis signal-regulated kinase 1 (ASK1), PI3 kinase (PI3K), protein tyrosine phosphatase (PTP), and Src homology 2 domain-containing (Shc)); antioxidant and anti-inflammatory response (thioredoxin (TRX), redox-factor 1 (Ref-1), and NFE2-like 2 (Nrf-2)); iron homeostasis (iron regulatory protein (IRP)); and DNA damage response (ataxia–telangiectasia mutated (ATM)).
In order to understand ROS regulation of signaling pathways, the mechanism of how ROS alters protein function should be briefly addressed. The oxidative interface consists mainly of the redox regulation of redox-reactive cysteine (Cys) residues on proteins by ROS. Oxidation of these residues forms reactive sulfenic acid (−SOH) that can form disulfide bonds with nearby cysteines (−S–S−) or undergo further oxidation to sulfinic (−SO2H) or sulfonic (−SO3H) acid; if nearby nitrogen is available sulfenic acid may also form a sulfenamide. These oxidative modifications result in changes in structure and/or function of the protein. With the exception of sulfonic acid, and to a lesser degree sulfinic acid, these redox modifications are reversible by reducing systems such as thioredoxin and peroxiredoxin [7] which are necessary given that these modifications function in redox sensing and signaling. For a more detailed overview of redox chemistry refer to Winterbourn [8] and Janssen-Heininger [9].
2. Regulation of MAPK signaling pathways by ROS
The mitogen-activated protein kinase (MAPK) cascades consist of four major MAPKs; the extracellular signal-related kinases (Erk1/2), the c-Jun N-terminal kinases (JNK), the p38 kinase (p38), and the big MAP kinase 1 (BMK1/Erk5). These kinases are evolutionarily conserved in eukaryotes and play pivotal roles in cellular responses to a wide variety of signals elicited by growth factors, hormones, and cytokines, in addition to genotoxic and oxidative stressors. The function and regulation of the MAPK cascades have been comprehensively covered [10-13]; therefore, this review article focuses solely on those protein kinases and phosphatases in the MAPK cascades that are directly regulated by ROS. MAPK pathways are composed of a three-rung kinase tier; MAPK kinase kinases (MAPKKK) phosphorylate and activate MAPK kinases (MAPKK), which in turn phosphorylate and activate MAPKs. Among the members of the MAPK cascades, apoptosis signal-regulated kinase 1 (ASK1) is an upstream MAPKKK that regulates the JNK and p38 MAPK pathways leading to apoptosis through phosphorylation of MKK4, MKK3, and MKK6 MAPKKs [14]. ASK1 is activated under various stress conditions including oxidative stress [15]. ASK1 is homo-oligomerized by both C- and N-terminal coiled-coil domain interaction and activation occurs through phosphorylation of a conserved threonine (Human: Thr-838, Mouse: Thr-845) residue in the activation loop of the human ASK1 kinase domain (Fig. 2A). There are many ASK1-associated proteins so far identified, among which the redox protein thioredoxin was shown to constitutively interact with ASK1 and directly inhibit its kinase activity [16]. Only the reduced form of thioredoxin interacts with ASK1; by blocking N-terminal, but not basal C-terminal interaction, thioredoxin inhibits complete ASK1 oligomerization and subsequent activation (Fig. 2A) [16,17]. ASK1 is activated when oxidants or ROS oxidize two cysteine residues in the redox center of thioredoxin, inducing formation of an intramolecular disulfide bond between Cys-32 and Cys-35 that results in the dissociation of thioredoxin from ASK1, allowing for the subsequent N-terminal homophilic interaction and complete oligomerization of ASK1 (Fig. 2A), which is enhanced by the binding of tumor necrosis factor-α receptor associated factors (TRAF) [16-18]. The ASK1 oligomer subsequently undergoes autophosphorylation of a conserved threonine residue (Human: Thr-838, Mouse: Thr-845) located in the activation loop of ASK1 (Fig. 2A) [19], which is inactivated by protein phosphatase 5 (PP5) [20]. In addition to homo-oligomerization of ASK1, ASK1 hetero-oligomerizes with ASK2, another ASK family serine/threonine MAPKKK (Fig. 2B). ASK2 binds to the C-terminal domain of ASK1, and this interaction stabilizes ASK2, resulting in autophosphorylation of ASK2 at the conserved threonine (Human: Thr-806, Mouse: Thr-807) in the activation loop. ASK1 is then phosphorylated at Thr-838 by ASK2, resulting in activation of the hetero-oligomer (Fig. 2B) [21]. ASK1-deficient mouse embryonic fibroblasts were shown to be less susceptible to TNF- or H2O2-induced cytotoxicity along with decreased JNK and p38 MAPK activation, suggesting that ASK1 plays a pivotal role in promoting cell death under oxidative stress [15]; however, ROS activated ASK1 mediates p38 signaling leading to non-apoptotic outcomes also, such as differentiation [22] and immune signaling [23], thus reinforcing the role of ROS signaling in cellular homeostasis.
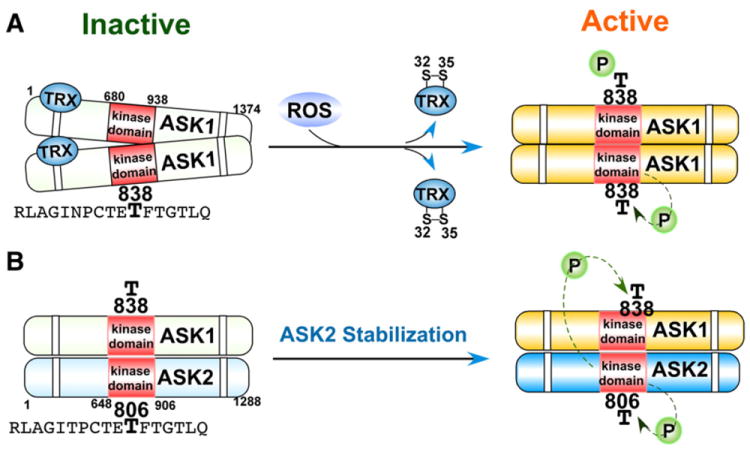
Activation of ASK kinases in response to oxidative stress. A) Oxidation of thioredoxin (TRX) results in disulfide bond formation between Cys-32 and Cys-35 and subsequent dissociation from ASK1. ASK1 undergoes complete homo-oligomerization and subsequent autophosphorylation at Thr-838 located in the kinase domain. B) Hetero-oligomerization of ASK1 and ASK2 stabilizes ASK2, resulting in 1) the autophosphorylation of ASK2 at Thr-806, and 2) the subsequent phosphorylation of ASK1 at Thr-838 by ASK2.
Evidence suggests that cGMP-dependent protein kinase (PKG) regulates MAPK activation [24,25]. It was demonstrated that PKG1α is a redox sensor activated by ROS; oxidation of Cys-42 by H2O2 resulted in the formation of an active PKG1α homodimer through intermolecular disulfide bond formation [26], though it is unknown whether ROS-activated PKG1α regulates MAPK pathways. Similarly, protein kinase A (PKA) was shown to be activated by ROS through formation of intramolecular disulfide bonds [27], and protein kinase C (PKC) activity is also regulated through redox mechanisms [28]; both kinases have been implicated in MAPK signaling.
MAPK pathways are also activated by the direct inhibition of MAPK phosphatases by ROS. ROS produced by NADPH oxidases or in mitochondria have been shown to inhibit JNK-inactivating phosphatases [29] through reversible oxidation of a catalytic-site cysteine to sulfenic acid, thus sustaining JNK activation. Inhibition of phosphatases by ROS has also been shown to regulate p38 signaling [30,31], and it was recently demonstrated that ROS generated by commensal bacteria inactivated dual-specific phosphatase 3 (DUSP3) by oxidation of Cys-124, resulting in ERK activation [32]. DUSP is a protein tyrosine phosphatase (PTP); inactivation of classical PTPs such as protein tyrosine phosphatase 1B (PTP1B) or SH2-domain containing PTP (SHP2) by ROS in a similar cysteine redox mechanism (I/V-H-C-X-X-G-X-X-R-S/T) [33-35] ultimately potentiates MAPK and growth factor signaling pathways initiated from RTK, cytokines, and stressors [33,35-41]. Oxidation of the catalytic site cysteine in PTPs to sulfenic acid (− SOH) is reversible, as are disulfide bonds and sulfenamides, but further oxidation to the generally irreversible sulfinic acid (−SO2H) or sulfonic acid (−SO3H) can also occur (Fig. 3). Thioredoxin or glutathione appears to be involved in reducing sulfenic acid residues and reversing the oxidative inactivation of PTPs [34]. The classical PTP family includes the ligand binding transmembrane receptor-like PTPs (RPTPs) such as RPTPα. Ligand binding to RPTP induces RPTP dimerization, which results in the catalytic-inactive conformation of RPTP [42]. ROS have been shown to inhibit human RPTPα tyrosine phosphatase activity through preferential oxidation of Cys-723 in the second catalytic domain of RPTPα rather than oxidation of Cys-433 in the first catalytic domain [43], leading to the formation of intermolecular Cys – Cys disulfide bonds as well as a reversible cyclic sulfenamide [44] and subsequent stabilization of the inactive RPTPα dimer [45]. Growth factor signaling events are frequently associated with production of ROS that are known to be important signaling molecules [46]. Oxidation and inhibition of PTPs by ROS appear to be one of the molecular mechanisms through which growth factor-induced ROS production is essential for transducing and sustaining growth factor signals.
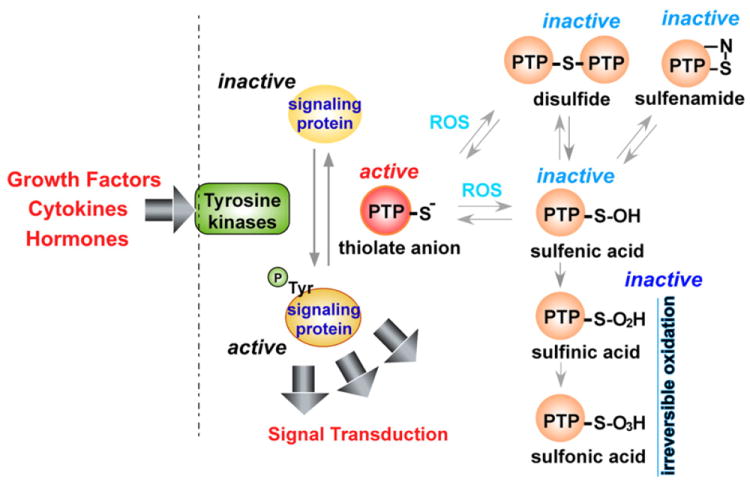
Mechanism of ROS-mediated protein tyrosine phosphatase inactivation. Tyrosine kinases, activated by growth factors, cytokines, and hormones, phosphorylate target proteins. Phosphorylation can be reversed by protein tyrosine phosphatases (PTP); ROS inactivates PTP by oxidation of catalytic cysteine residues resulting in the formation of the sulfenic acid (−SOH) intermediate that can form disulfide bonds or sulfenamide residues. Further oxidation of sulfenic acid results in formation of sulfinic (−SO2H) or sulfonic acid (−SO3H), which are relatively irreversible.
3. Regulation of PI3K signaling pathways by ROS
Another signaling pathway that plays a key role in cell proliferation and survival in response to growth factor, hormone, and cytokine stimulation is the phosphoinositide 3-kinase (PI3K) pathway. The PI3K, consisting of the p110 catalytic subunit and the p85 regulatory subunit, is tightly coupled with RTKs activated by various growth factors such as Epidermal Growth Factor (EGF), Platelet-Derived Growth Factor (PDGF), Nerve Growth Factor (NGF), insulin, and Vascular Endothelial Growth Factor (VEGF). PI3K is recruited to activate tyrosine-phosphorylated RTK dimers through a SH2 domain in the PI3K p85 regulatory subunit. PI3K catalyzes the synthesis of the second messenger phosphatidylinositol 3,4,5 triphosphate (PIP3) from phosphatidylinositol 4,5 bisphosphate (PIP2), wherein the membrane bound PIP3 serves as a signaling molecule to recruit proteins containing the pleckstrin homology (PH) domain. These PH domain proteins, such as the phosphoinositide-dependent protein kinase (PDK) and protein kinase B (AKT) serine/threonine kinases, are thus activated and mediate further downstream signaling events [47]. The synthesis of PIP3 is negatively regulated primarily by the phosphatase and tensin homology (PTEN) phosphatase, which dephosphorylates PIP3 back to PIP2 [48]. Through PTEN, the PI3K pathway is subject to reversible redox regulation by ROS generated by growth factor stimulation. H2O2 was shown to oxidize and inactivate human PTEN through disulfide bond formation between the catalytic domain Cys-124 and Cys-71 residues [49,50]. It was also demonstrated that endogenously generated ROS following treatment with peptide growth factors such as insulin, EGF, or PDGF causes oxidation of PTEN leading to the activation of the PI3K pathway [51]. PTEN oxidation is reversed by peroxiredoxin II, a cytoplasmic peroxiredoxin isoform that eliminates H2O2 generated in response to growth factors [49]. Thus the PI3K pathway is regulated by ROS in a similar manner as the MAPK pathways; at the oxidative interface, protein phosphatases are directly oxidized by ROS resulting in sustained activation of the signaling pathways. It is noteworthy that various oxidants and ROS-producing chemicals activate transcription of a battery of antioxidant genes through a PI3K-NFE2-like 2 (Nrf2)-antioxidant response element (ARE) mechanism (Section 4), where PTEN knockdown enhances transcription of ARE-regulated antioxidant genes [52]; however, it is not known whether these oxidants induce PTEN oxidation and inhibition of phosphatase activity leading to gene activation.
4. Nrf2 and Ref1-mediated redox cellular signaling
In order to prevent oxidative stress, the cell must respond to ROS by mounting an antioxidant defense system. Antioxidant enzymes play a major role in reducing ROS levels; therefore, redox regulation of transcription factors is significant in determining gene expression profile and cellular response to oxidative stress. Redox factor-1 (Ref-1) (Fig. 4A), identified as a 37-kDa protein that facilitates Fos-Jun DNA binding activity [53], was shown to be identical to an apurinic/apyrimidinic (AP)-endonuclease named APE (AP endonuclease) [54] or human AP endonuclease 1 (HAP1) [55]. Thus Ref-1 is a multi-functional protein that not only regulates transcription factor activity, but also mediates base excision repair. The transcriptional regulatory function of Ref-1 is mediated through its redox activity on several transcription factors such as activator protein 1 (AP-1), p53, nuclear factor kappa B (NFkB), and hypoxia inducible factor 1 (HIF-1α) [56]. The N-terminus region of Ref-1 is responsible for redox activity while the AP-endonuclease activity domain is located at the C-terminal region (Fig. 4A) [57]. Cys-65 of Ref-1 appears to be a major redox active site (along with Cys-93) that is required for the reduction and increased DNA binding of targeted transcription factors [58]. Ref-1 activated the AP-1 transcription factor, Fos-Jun, through redox regulation of cysteine residues in the Fos-Jun DNA binding domains [53,59,60]. As shown in Fig. 4B, this cysteine is highly conserved in various human b-zip transcription factors, and except for CAATT enhancer binding protein (C/EBP) transcription factors, all may be regulated in a redox dependent manner by Ref-1, resulting in increased DNA binding and transcriptional activation of target genes. Indeed, it was demonstrated that this conserved cysteine in Nrf2 and cAMP response element binding (CREB) protein is subject to redox regulation. Site mutagenesis of Cys-506 interfered with mouse Nrf2-antioxidant response element (ARE) binding [61], in contrast to mutation of Cys-300/310 of CREB which increased DNA binding activity [61,62], demonstrating the importance of these redox regulated cysteine residues in transcriptional activity. Furthermore, Ref-1 was shown to be involved in the transcriptional activation of Nrf2-target genes under oxidative stress [63]. Oxidation and inactivation of b-zip transcription factors is not due to formation of intra-or intermolecular disulfide bonds, but probably the result of reversible oxidation of cysteine to sulfenic (− SOH) or sulfinic (− SO2H) acids [59], though the possibility of an intermolecular disulfide bond between Cys-300 and Cys-310 in CREB cannot be entirely discounted [62]. It is generally accepted that reduction of Ref-1-targeted transcription factors results in oxidation of Ref-1 at Cys-65, possibly along with Cys-93 (Fig. 4A), although it has not been completely determined whether this oxidation event results in the formation of an intramolecular disulfide bond or the conversion to sulfenic or sulfinic acid [64].
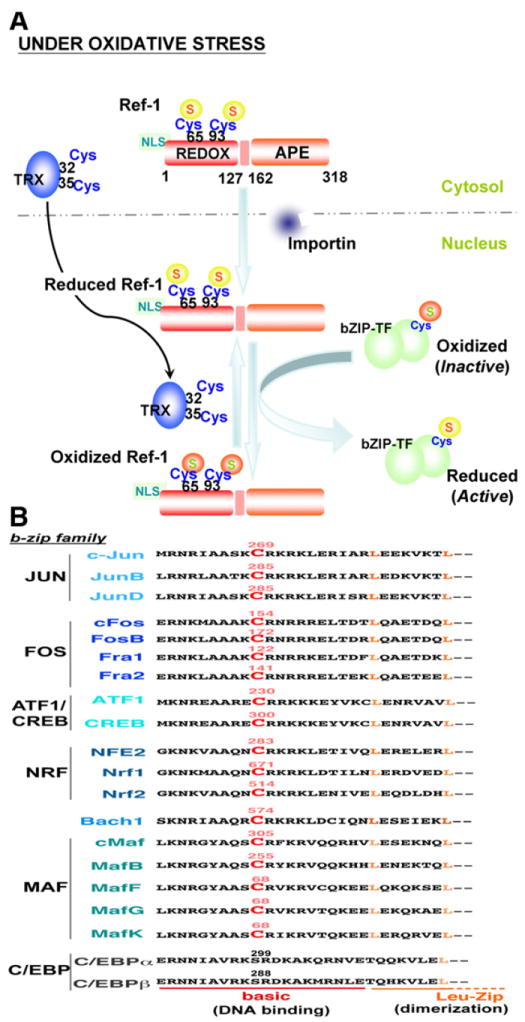
Ref1-mediated redox cell signaling. A) The human Ref-1N-terminal region consists of the redox domain (REDOX; residues 1–127) and a 20 amino acid nuclear localization sequence (NLS); the C-terminal region contains the apurinic/apyrimidinic endonuclease domain (APE; residues 162–318). Cysteine-65 and -93 are major redox active sites. Under oxidative stress, Ref-1 translocates into the nucleus by NLS-importin-dependent or -independent pathways where Ref-1 regulates the activity of b-zip transcription factors (bZIP-TF) by redox mechanisms. Oxidized Ref-1 is subject to redox regulation by nuclear translocated thioredoxin (TRX) through the TRX catalytic center (Cys-32 and -35). B) The conserved redox-active cysteine of various human b-zip transcription factors is indicated in red color and labeled with the residue number. In addition to the b-zip family, other transcription factors such as p53, NFκB, and HIF-1α are also regulated by Ref-1 [56].
The reduction of Ref-1 appears to be regulated by thioredoxin (Fig. 4A). In response to phorbol myristate acetate or ionizing radiation, thioredoxin was shown to translocate into the nucleus and interact with Ref-1, resulting in the activation of AP-1 transcriptional activity under reducing conditions [65,66]. The interaction of thiore-doxin with Ref-1 and the subsequent activation of Ref-1 target proteins appear to be regulated by the redox active Cys-32 and Cys-35 residues of thioredoxin [65-67] which are responsible for its reducing activity [68] (Fig. 4A). Ref-1 is localized in the nucleus as well as in the cytoplasm, depending on the cell type and physiological conditions. Cytoplasmic Ref-1 translocates to the nucleus under oxidative stress conditions such as H2O2 [69] and hypochlorous acid (HOCl) [70]. It was demonstrated that nuclear importins interact with the N-terminal 20 amino acid region of Ref-1, mediating nuclear translocation (Fig. 4A) [71]. It remains unknown how oxidants or ROS trigger nuclear localization of Ref-1, though it has been shown that nitric oxide induces nuclear export of Ref-1 by S-nitrosation of the redox sensitive Cys-93 residue [72]; p38 may also play a role in the nuclear translocation of Ref-1 via phosphorylation of Ser-54 [73]. These reports suggest that Ref-1 nuclear translocation by oxidants or oxidative stress responsive signaling pathways may occur through modifications of regions outside the accepted nuclear localization sequence of Ref-1.
Increasing the cellular antioxidant capacity by upregulation of antioxidant detoxification genes is critical in cellular adaptation to oxidative stress and protection from oxidative damage. Ref-1 was shown to be upregulated by genotoxic agents and oxidants, such as bleomycin and H2O2, and so protected cells from DNA and oxidative damage [70]. Ref-1 upregulation seems to be a reasonable adaptive response since Ref-1 mediates both DNA repair and the redox activation of key transcription factors involved in cellular defense, such as AP-1 and NFkB. However, under oxidative stress conditions, a group of antioxidant detoxification genes such as glutathione S-transferase (GST) [74], NADPH quinone oxidoreductase-1 (NQO1) [75], heme oxygenase-1 (HO1) [76,77], and ferritin H (FH) [78,79] are transcriptionally activated in an AP-1/NFkB-independent manner. These antioxidant genes are regulated by a highly homologous enhancer termed the antioxidant responsive element (ARE), or electrophile response element (EpRE), located ~0.5 kb–10 Kb upstream from transcription initiation sites of these genes [74-76,78-80]. The consensus core ARE sequence is TGA(C/T)nnnGCA [81] and the presence of two or more copies of the ARE in close proximity to each other often serves as a bona fide ARE [80]. Various endogenous and exogenous ROS-generating and electrophilic chemicals (such as H2O2 [82], lipid aldehydes [83], arsenic [84], tert-butylhydroquinone (t-BHQ) [82], hemin [63,82,85], and resveratrol [86]) activate transcription of these antioxidant genes via the ARE.
The primary transcription factor involved in ARE activation under oxidative stress conditions is NFE2-like 2 (Nrf2), a cap ‘n’ collar (CNC)-b-zip transcription factor (Fig. 5) [87]. Under non-stressed conditions, the majority of Nrf2 resides in the cytoplasm (though there is controversy as to the exact subcellular localization of Nrf2 [88]) and associates with a dimeric inhibitory protein, Kelch-like ECH-associated protein-1 (Keap1) [87]; Keap1 interacts with the cullin-3 E3-ubiquitin ligase (Cul3) and serves as a platform for the ubiquitination and resultant proteasomal degradation of Nrf2 (Fig. 5) [89,90]. Reactive mouse Keap1 cysteines (Cys-151, -273, and -288) [91,92] are redox sensors, and upon oxidation by ROS, results in the dissociation of Nrf2 from Keap1/Cul3 which allows Nrf2 stabilization and translocation into the nucleus. In the nucleus Nrf2 dimerizes with members of another b-zip family, the small Maf proteins (Maf-F, Maf-G, and Maf-K). The Nrf2-small Maf heterodimer binds the ARE enhancer and activates ARE-dependent transcription of target genes which serve as antioxidants and in processes such as electrophile detoxification, glutathione synthesis, and ROS homeostasis (Fig. 5) [80,93]. As shown in Fig. 4B, Nrf2 contains a conserved cysteine located in the DNA binding domain (Cys-514) which is the conserved site of Ref-1-mediated redox regulation. Indeed, mutation of mouse Nrf2 at Cys-506 (equivalent to Cys-514 of the human Nrf2) to Ser-506 affected binding to the ARE enhancer and decreased NQO1 expression [61]. It has also been demonstrated that Ref-1 nuclear localization and transcriptional activation of the ARE in the human ferritin H gene were increased following t-BHQ or hemin treatment [63], suggesting the possibility of Nrf2 redox regulation by Ref-1. Another example of possible redox regulation of Nrf2 was shown when mutation of the Cys-119 residue located in the transactivation domain of Nrf2 evidenced decreased binding to the NQO1 ARE [94].
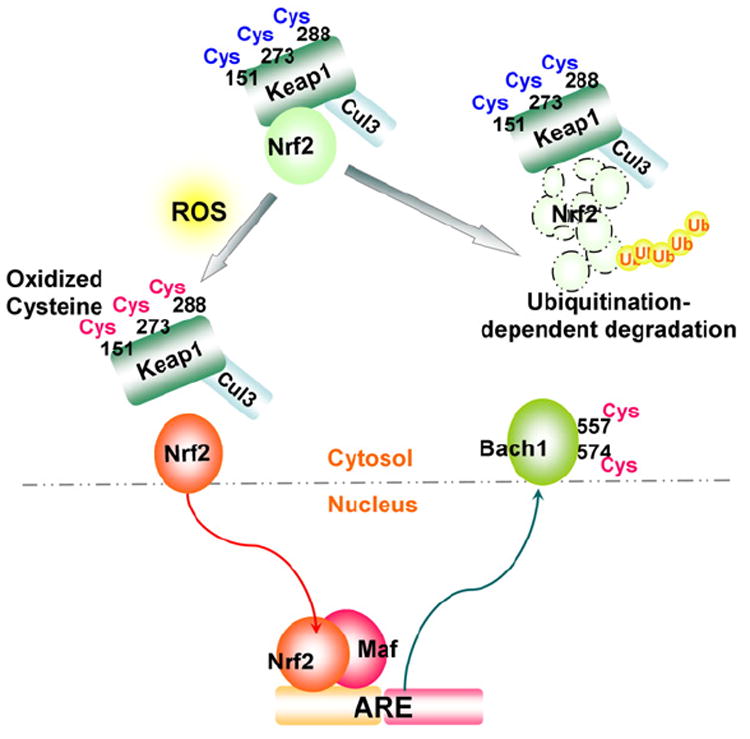
Redox regulation of the Nrf2–ARE pathway. ROS oxidation of cysteines (Cys-151, -273 and -288) in the mouse Kelch-like ECH-associated protein-1 (Keap1) results in the release of Nrf2 from the Keap1/cullin-3 E3-ubiquitin ligase (Cul3) complex, preventing Nrf2 degradation. Nrf2 subsequently undergoes nuclear translocation. In the nucleus, a heterodimer of Nrf2 and small Maf members (Maf-F, Maf-G, and Maf-K) binds the antioxidant-responsive element (ARE); oxidation of a b-zip transcriptional repressor of ARE, the human BTB and CNC homolog 1 (Bach1) at Cys-557 and -574 results in cytoplasmic translocation of Bach1, both leading to activation of the ARE.
Interestingly, the BTB and CNC homolog 1 (Bach1), a b-zip transcriptional repressor of the ARE [95], also features the Ref-1 associated conserved cysteine (Cys-574) (Fig. 4B) that is subject to redox regulation [96]. In this study, the sulfhydryl oxidizing agent diamide reversed Bach1-repressed ARE enhancer activity via Cys-574 oxidation (probably Cys-557 as well) leading to cytoplasmic translocation of Bach1 (Fig. 5) [96]. Nuclear export of Bach1 during ARE-dependent transcriptional activation of the NQO1 gene after t-BHQ treatment is also facilitated through phosphorylation of the mouse Bach1 at Tyr-486 (Tyr-483 in the human Bach1) by an undetermined tyrosine kinase [97]. Collectively, these results suggest that at least two sequential redox events, 1) the oxidation or adduct formation of Keap 1 in the cytoplasm and subsequent release and nuclear translocation of Nrf2, and 2) the redox regulation of Nrf2 and Bach1 in the nucleus, appear to be critical for maximum transcriptional activation of ARE-dependent antioxidant genes via the Nrf2 signaling pathway. Thus, through upstream redox regulators such as Ref-1, transcription factor and repressor activity is modulated indirectly through ROS, while the examples of Nrf2 and Bach1 demonstrate the direct regulation of transcription factors and repressors by ROS. Both direct and indirect control of transcriptional regulators illuminate the oxidative interface between ROS and ARE gene transcription.
5. Regulation of p66Shc, mitochondrial oxidative stress, and aging
ROS have been implicated in the process of aging. Given that the majority of endogenous ROS are generated in mitochondria [98], there has been much interest in the role that mitochondrial ROS may play in aging. Of note is the Shc adaptor protein family, encoded by the shcA locus in mammalian cells, consisting of the p66Shc, p52Shc, and p46Shc isoforms (Fig. 6) [99-101]. Expression of p66Shc and p52/p46Shc isoforms is regulated by two different promoters [102] along with alternative translation initiation or splicing [99]. All isoforms share a phosphotyrosine binding domain (PTB), and a proline-rich collagen homology domain-1 (CH1) followed by a C-terminal Src homology 2 domain (SH2) (Fig. 6). p52Shc and p66Shc also share a cytochrome c binding domain (CB), while a second CH domain (CH2) is unique to pShc66 (Fig. 6). The homozygous shcA gene knockout mice lacking shcA exons 2 and 3 are embryonically lethal by E11.5 due to congestion of blood in the heart and cardiac outflow tracts [103]. The predominant expression of Shc proteins in the developing cardiovascular system indicates the importance of Shc proteins in the development of the heart and angiogenesis [103]. Among the three Shc isoforms, p52Shc and p46Shc are adaptor proteins involved in RTK signaling pathways through recruitment of the SH2 domain to phospho-tyrosines in the cytoplasmic domain of RTKs upon growth factor stimulation [99,103]. In contrast, p66Shc was revealed to play more predominant roles in mitochondrial ROS metabolism and oxidative stress response rather than serving as an RTK adaptor protein. Pelicci and colleagues demonstrated that p66Shc-deficient mice are not only more resistant to apoptosis under oxidative stress, but also have increased life span [104]. p66Shc-deficient mouse fibroblasts also showed decreased toxicity in response to oxidative stress compared with normal p66Shc fibro-blasts and in vitro results suggest that phosphorylation of p66Shc at Ser36 is critical for stress-induced apoptosis [104]. Subsequently, a fraction of p66Shc was shown to localize in mitochondria as a high molecular mass protein complex containing heat shock protein 70 (HSP70), and a modest increase in mitochondrial p66Shc along with its dissociation from this large protein complex were observed after UVC or H2O2 exposure [105]. p66Shc mitochondrial translocation or proapoptotic activity may be regulated by posttranslational modifications such as phosphorylation of p66Shc at Ser-36 in the CH2 domain (Fig. 6) by PKC or JNK following exposure to UV or H2O2, and/or interaction with TOM–TIM protein import complexes [105-109], although it was previously noted that p66Shc in mitochondria is not serine phosphorylated [110].
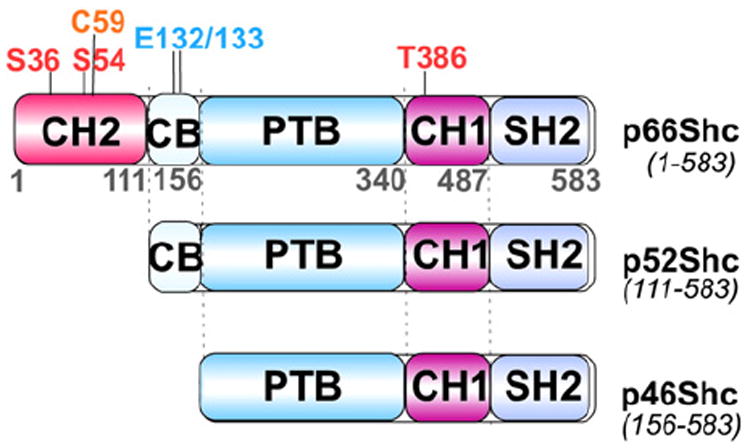
Isoforms of p66Shc, p52Shc and p46Shc. Schematic representation of three isoforms produced from the human ShcA gene (CH1 and 2, proline-rich collagen homology domain-1 and 2; CB, cytochrome c binding domain; PTB, phosphotyrosine binding domain; and SH2, Src homology 2 domain); numbers define the regions of different domains according to human p66Shc amino acid sequence; Met-111 and -156 of p66shc are equivalent to the first methionines of p52Shc and p42Shc. ROS-mediated phosphorylation sites are at Ser-36 and -54; Thr-386; pro-apoptotic residues are Cys-59 and Glu-132/133.
Our current understanding is that p66Shc is a proapoptotic protein involved in ROS production in mitochondria leading to mitochondrial damage and apoptosis under oxidative or genotoxic stress conditions such as H2O2 or UV exposure. What is the initial and direct impact of oxidative or genotoxic stress on p66Shc and how is p66Shc activated? First, p66Shc protein levels in cytoplasm as well as in mitochondria appear to be increased under certain stress conditions [111,112] in addition to increased serine and threonine phosphorylation of p66Shc [100,101]. Although the molecular mechanism by which p66Shc expression is increased in response to stress signals remains largely uncharacterized, the Rac1 GTPase, which generates ROS through activation of NADPH oxidase [113], was shown to block p66Shc ubiquitination and degradation through phosphorylation of p66shc on Ser-54 and Thr-386 in a p38 dependent manner [114]. Furthermore, oxidative stress-activated PKC-β induces phosphorylation of p66Shc at Ser-36, which in turn triggers the interaction of the prolyl isomerase Pin1 with p66Shc, possibly inducing the isomerization of a p66shc phospho-Ser36-Pro37 bond, resulting in the subsequent translocation of p66Shc into mitochondria [115]. Secondly, p66Shc was shown to be pro-apoptotic in mitochondria upon redox-dependent reversible tetramerization through formation of two disulfide bonds via Cys-59 in the N-terminus CH2 domain of p66Shc (Fig. 6), leading to copper-dependent ROS generation and initiation of apoptosis [106]. The active oxidized form of p66Shc is reversibly reduced by glutathione or thioredoxin leading to inactivation [106]. The molecular mechanism through which stress-activated p66Shc induces apoptosis has not been fully elucidated; however, p66Shc was shown to serve as a redox protein that produces H2O2 in mitochondria through interaction and electron transfer between p66Shc and cytochrome c [110], in which mutations in the redox center of p66Shc (E132–E133 to Q132–Q133 in the CB domain, Fig. 6) impaired opening of the mitochondrial permeability transition pore and thus negating the proapoptotic function of p66Shc [99]. It appears that p66shc induces apoptosis through generation of ROS but also may be activated by ROS. Aging is associated with a decrease in mitochondrial function such as impaired oxidative phosphorylation that results in increased generation of ROS [116]. It is interesting to speculate whether increased ROS in this context would activate p66shc to produce further ROS, resulting in apoptosis and maintaining the steady progression of aging. Regardless, p66shc remains an interesting link between ROS and aging.
6. Regulation of the IRE–IRP system and iron homeostasis by ROS
Iron is an essential element that plays crucial roles in cell proliferation and metabolism by serving as a functional constituent of various enzymes including ribonucleotide reductase and cytochrome P450. However, when present in excess, free iron generates ROS via the Fenton reaction [117-119], placing cells under deleterious oxidative stress. Therefore, tight regulation of iron homeostasis is crucial not only to maintain normal cellular function, but also to prevent iron-mediated oxidative stress. The expression of many genes involved in iron transport and storage is regulated by iron itself at the post-transcriptional level in which iron regulatory protein-1 and -2 (IRP1 and IRP2) interact with an iron-responsive element (IRE) in the 5′-or 3′-untranslated region (UTR) of mRNAs such as, for example, ferritin (serving as intracellular iron storage) and transferrin receptor-1 (serving as iron transport into cells), respectively (Fig. 7). The IREs consist of a stem-loop structure composed of approximately 30 nucleotides with a 5′-CAGUG-3′ loop to which cytoplasmic IRP1 and/or IRP2 binds and determines the fate of mRNAs. In general, IRP binding to IRE in the 5′-UTR of mRNA (e.g. ferritin) results in mRNA translational block and decreased protein expression while IRP binding to the 3′-UTR of mRNA (e.g. transferrin receptor-1) increases the stability of mRNA, increasing protein expression [117,120]. The amino acid sequence of IRP1 is highly homologous to mitochondrial aconitase, and IRP exhibits aconitase activity when forming a 4Fe–4S iron–sulfur cluster [121,122] and it is through this cluster that cellular iron levels control IRP1–IRE interaction. Under iron-rich conditions IRP1 retains the 4Fe–4S cluster (inactive IRP1) and therefore cannot bind to the IRE (Fig. 7). IRP2, on the other hand, is highly homologous to IRP1 but lacks an iron–sulfur cluster and has no aconitase activity. Furthermore, under iron rich conditions IRP2 is degraded via the proteasome pathway (Fig. 7). Thus, iron-rich conditions induce dissociation of IRPs from IREs, resulting in release of the translational block of ferritin mRNA and the destabilization of transferrin receptor 1 mRNA as well. As a result, iron-rich cells synthesize more ferritin for iron storage/detoxification and less transferrin receptor-1 to halt iron transport into the cells, ultimately reducing excess intracellular iron. In contrast, iron-deficient conditions facilitate the disassembly of the 4Fe–4S cluster in IRP1 while stabilizing IRP2, allowing IRP1 and IRP2 to bind IREs in 5′-UTR ferritin mRNA (translational block) and 3′-UTR transferrin receptor-1 mRNAs (mRNA stabilization). As a result, iron-deficient cells produce less ferritin (decreasing iron storage capacity) and more transferrin receptor-1 (increasing iron transport) to maintain iron homeostasis [117,120]. In the last decade several new iron transport and metabolism genes such as Divalent Metal Transporter 1 (DMT1) and ferroportin (Fpn) [117] were discovered. The identification of IREs present in the 5′- or 3′-UTR of Fpn and DMT1 mRNAs strengthens the view of the IRE–IRP regulatory system as the primary post-transcriptional mechanism of the majority of iron metabolism genes tightly regulated by iron (Fig. 7). In addition, other genes are regulated by the IRE–IRP system, such as NADH dehydrogenase (ubiquinone) Fe–S protein 1 (NDUFS1) [123], Alzheimer’s amyloid precursor protein (APP) [124], hydroxyacid oxidase 1 (HAO-1) [125], myotonic dystrophy kinase-related Cdc42-binding kinase alpha (MRCKα) [126], cell division cycle 14 homolog A (CDC14A) [127], delta-aminolevulinate synthase 2 (ALAS2) [128], and hypoxia inducible factor-2 alpha (HIF-2α) [129] (Fig. 7).
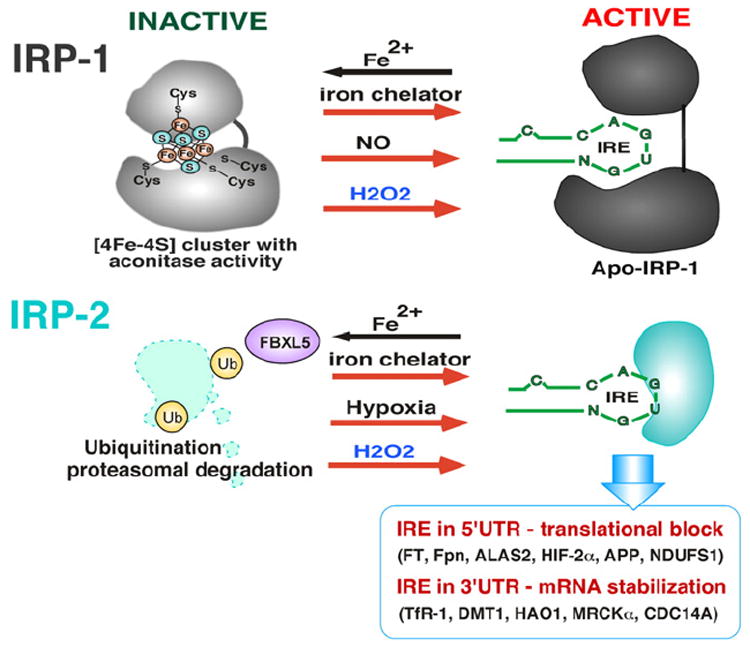
Regulation of IRP–IRE interactions. Under iron rich conditions, IRP1 contains a [4Fe–4S] cluster and is unable to bind to the IRE, though loss of iron from the cluster (destabilization) under iron deficient conditions allows IRP1 to bind to the IRE. IRP2 does not contain a [4Fe–4S] cluster and is degraded by F-box/LRR-repeat protein 5 (FBXL5)-dependent ubiquitination. Iron chelators, nitric oxide (NO), hypoxia, and hydrogen peroxide (H2O2) increase IRP/IRE interaction. H2O2 destabilizes the [4Fe–4S] cluster of IRP1 and also stabilizes IRP2 protein by preventing FBXL5-dependent ubiquitination. Increasing IRP–IRE interaction in 5′UTR results in translational block of ferritin (Ft), ferroportin (Fpn), aminolevulinic acid synthase-2 (ALAS2), hypoxia inducible factor-2α (HIF-2α), amyloid precursor protein (APP), and NADH dehydrogenase (ubiquinone) Fe–S protein 1 (NDUFS1) genes, but in the 3′UTR it results in mRNA stabilization of transferrin receptor (TfR), divalent metal transporter 1 (DMT1), hydroxyacid oxidase 1 (HAO-1), myotonic dystrophy kinase-related Cdc42-binding kinase alpha (MRCKα), and CDC14 cell division cycle 14 homolog A (CDC14A).
The fact that IRP1 contains a 4Fe–4S cluster implies that it may be subject to redox regulation. Indeed, H2O2 was shown to convert, or destabilize, the 4Fe–4S cluster of IRP1 (inactive) to a 3Fe–4S cluster (active) (Fig. 7) through loss of a single iron [122], and ferritin protein expression was transiently downregulated after H2O2 exposure [78,130] through increased IRP1 binding to the IRE, though followed by upregulation of ferritin by transcriptional activation of the ferritin gene via the ARE [78]. The question that arises is whether increased IRP1–IRE binding is the direct effect of H2O2 on the 4Fe–4S cluster. When IRP1 was directly incubated with H2O2 in vitro, there was no increase in IRP1 binding to IRE [122,130], suggesting that destabilization of the 4Fe–4S cluster is not sufficient for IRP1 binding to IREs in response to H2O2, implying that H2O2 activates an alternate signaling pathway leading to additional posttranslational modifications of IRP1 for increased IRE binding. Nitric oxide (NO) was also found to increase IRP1 binding to the IRE through destabilization of the 4Fe–4S cluster [131,132] (Fig. 7). The redox-regulated PKC was shown to phosphorylate IRP1 at Ser-138 [133], and it was later demonstrated that phosphorylation of Ser-138 results in destabilization of the 4Fe–4S cluster and increases IRP1 binding to IRE [134]. Unlike IRP1, IRP2 does not contain a Fe–S cluster and its binding to IREs is primarily decreased in iron-rich cells through iron-dependent proteasomal degradation mediated by F-box/LRR repeat protein 5 (FBXL5) [135] (Fig. 7), resulting in downregulation of iron transporting proteins (destabilization of such mRNAs as TfR1 and DMT1) and upregulation of iron storage and export proteins (release of translational block of such mRNAs as ferritin and ferroportin) [136]. It has recently been demonstrated that IRP2 is subject to redox regulation, in which oxidative stress caused by glucose deprivation in HEK293 cells induced oxidation of Cys-512 and Cys-516 in IRP2 that in turn decreased IRP2 binding to IREs [137], and decreased IRE binding ability of IRP2 was correlated with decreased transferrin receptor-1 expression that may allow cells to limit iron transport and hindering subsequent iron-mediated ROS production. However, in contrast, a recent report demonstrated that ROS increased IRP2–IRE binding [138] in addition to protecting IRP2 from iron-mediated degradation, an effect similar to that shown under hypoxic conditions [139] (Fig. 7).
Taken together, the IRE–IRP regulatory system is not only regulated by cellular iron status but also regulated by ROS, in which cells elicit a defense mechanism against iron toxicity and iron-catalyzed oxidative stress.
7. ROS and DNA-damage response
Ataxia–telangiectasia mutated (ATM) and Ataxia–telangiectasia and Rad3-related (ATR) are PI3K-like serine/threonine protein kinases activated under genotoxic stress conditions and phosphorylate various proteins involved in cell proliferation, cell death and survival, and DNA repair [140,141]. These two signaling proteins were initially thought to be activated by a particular type of DNA damage therefore serving in parallel signaling pathways; however, accumulating evidence suggests that the ATM- and ATR-pathways communicate and cooperate in response to DNA damage [141]. ATM, preferentially activated by DNA double strand breaks, has been shown to serve as a sensor of oxidative stress in which ATM-deficient cells were more susceptible to oxidative stress-inducing agents as well as DNA damaging agents [142]. However, it has recently been demonstrated that the molecular mechanisms of the activation of ATM by DNA damage and oxidative stress are different. Upon double strand DNA break induction by agents such as bleomycin, cells recruit the Mre11–Rad50–Nbs1 (MRN) complex to damaged sites together with ATM, which in turn triggers autophosphorylation of ATM at Ser-1981 and activates ATM protein kinase activity leading to phosphorylation of downstream signaling proteins such as checkpoint kinase 2 (Chk2) at Thr-68 and p53 at Ser-15 (Fig. 8). Phosphorylation of ATM at Ser-1981 and its kinase activity are reversibly regulated by protein phosphatase 2A (PP2A)[143]. Cells exposed to H2O2 also feature ATM activated via Ser-1981 phosphorylation [144-146], although Guo et al. showed that H2O2 activates ATM in an MRN/Ser-1981 autophosphorylation-independent manner (Fig. 8) based on their results that 1) ATM was activated by H2O2 equivalently in both wild type and mutant Mre11 cells, and 2) H2O2 activated both wild type and Ser-1981 to alanine mutant purified dimeric ATM in vitro [144]. Noncovalently associated dimeric (non-active) ATM is known to be dissociated into active monomers in response to DNA damage; however, Guo et al. showed that purified ATM protein incubated with H2O2 in vitro migrated slower in SDS-PAGE due to formation of covalent dimers that were sensitive to reducing agents, and given the fact that N-acetyl-cysteine (NAC) blocked ATM activation induced by H2O2 in vitro [144], these results suggest that H2O2 activates ATM through formation of active ATM dimers via intermolecular disulfide bond(s). Further characterization demonstrated that Cys-2991, located near the kinase domain of human ATM, is primarily involved in the disulfide bond formation and oxidative activation of ATM (Fig. 8) [144]. It is noteworthy that a C2991A ATM mutant was fully activated by the MRN–DNA complex but not by H2O2 in vitro [144]. Thus H2O2, and possibly other ROS, elicit ATM activation not through the DNA damage and MRN mediated pathway, but directly by ATM dimer formation via Cys-2991 oxidation and intermolecular disulfide bridge formation (Fig. 8).
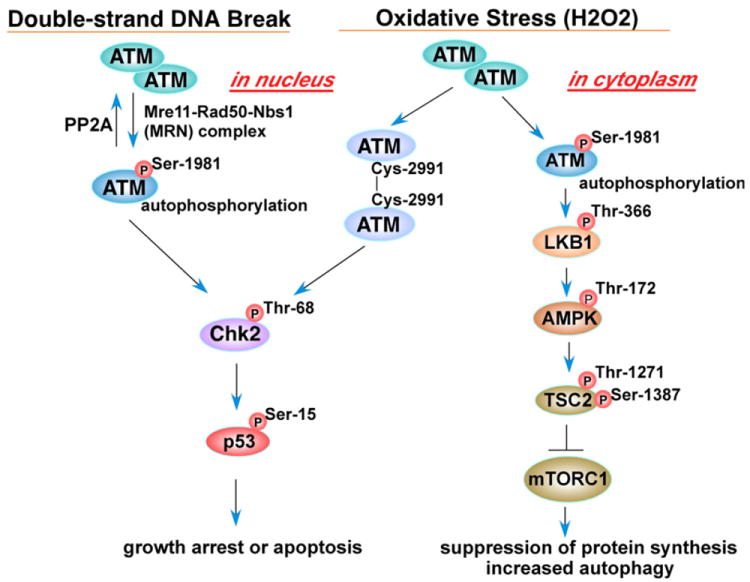
Schematic of ATM signaling upon oxidative stress and double-strand DNA breaks. Double-strand DNA breaks mediate the phospho-ATM–Chk2 pathway; however, oxidative stress elicits both the phospho-ATM–LKB1 and the ATM–homodimer pathways (ataxia–telangiectasia mutated (ATM); liver kinase B1 (LKB1); AMP activated protein kinase (AMPK); tuberous sclerosis complex 2 (TSC2); mammalian target of rapamycin complex1 (mTORC1)).
The fact that ATM deficient cells accumulate ROS and are sensitive to oxidative damage [142] suggests that ATM is crucial to a cellular oxidative stress defense program. What downstream signaling events are regulated by activated ATM in response to ROS? One clue to address this question has recently been presented [145] in which cytoplasmic ATM autophosphorylated at Ser-1981 in response to oxidative stress activates a liver kinase B1 (LKB1)-AMP activated protein kinase (AMPK) cascade (Fig. 8). Autophosphorylated cytoplasmic ATM activates LKB1 via phosphorylation of Thr-366, which activates AMPK through Thr-172 phosphorylation. Activated AMPK in turn activates the tuberous sclerosis complex 2 (TSC2) tumor suppressor protein via phosphorylation at Thr-1271 and Ser-1387, leading to inhibition of mammalian target of rapamycin complex 1 (mTORC1), thereby suppressing protein synthesis and inducing autophagy under oxidative stress (Fig. 8) [145]. The activation of autophagy through this pathway may be a cellular defense mechanism in response to ROS.
8. Conclusions
The disease states in which ROS signaling and toxicity have been implicated are areas of intensive research in regards to prevention and therapy. Unveiling the molecular mechanisms of disease pathogenesis and progression is therefore essential in providing relevant targets in order to develop innovative therapeutic strategies. In this context it is worthwhile not only to investigate ROS signaling in disease, but also to reveal how ROS instigate cellular signaling under homeostatic conditions. Having a clear understanding of how ROS directly regulate signaling pathways that are found to play a key role in the pathogenesis and progression of disease will allow us to understand how ROS may cause or contribute to disease and uncover new therapeutic targets. For example, ROS regulates proliferative and apoptotic pathways, and aberrant regulation of proliferation and apoptosis is essential in tumorigenesis; therapeutic strategies exploiting the role of ROS in those pathways are being developed [1,147]. While therapeutic development has primarily been concerned with reducing ROS levels to prevent toxicity, as in neurodegeneration [148], atherosclerosis, and diabetes [149], it remains to be seen whether targeting the redox sensitive molecules and signaling pathways activated by ROS will produce viable therapeutics in the prevention and alleviation of ROS-mediated disease states.
Content retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3454471/.